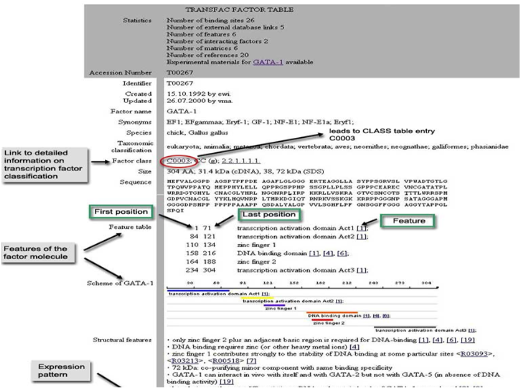
표피성장인자수용체(EGFR)의 증폭과 관련이 있는 질병 찾기
- Posted at 2010/10/11 09:09
- Filed under 제품소식
How can I find diseases that are involved in the amplification of a specific gene?
표피성장인자수용체(EGFR)의 증폭과 관련이 있는 질병에는 어떠한 것들이 있을까?
보통 표피성장인자수용체(EGFR)가 증폭되었을 때 어떤 질병과 관련이 있는지 알아보기 위해서 먼저 인터넷 또는 논문 검색을 하게된다. 인터넷 또는 논문 검색을 통해 찾은 정보들은 무수히 많고 그것을 하나하나 살펴보고 정리하는 것도 만만치 않은 일이다. 그래서 표피성장인자수용체가 증폭되었을 때 생길 수 있는 질병만 선택해서 찾고 한번에 Pathway까지 그릴 수 있는 방법을 소개하고자 한다.
Step to follow
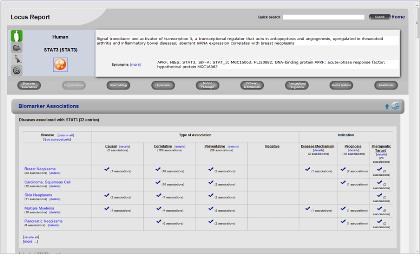
Step 1. 표피성장인자수용체(EGFR) 검색
PathwayStudio 검색창에서 표피성장인자수용체인 EGFR을 검색한다. 검색된 EGFR을 복사한 후 새 Pathway 문서에 붙여넣기 한다.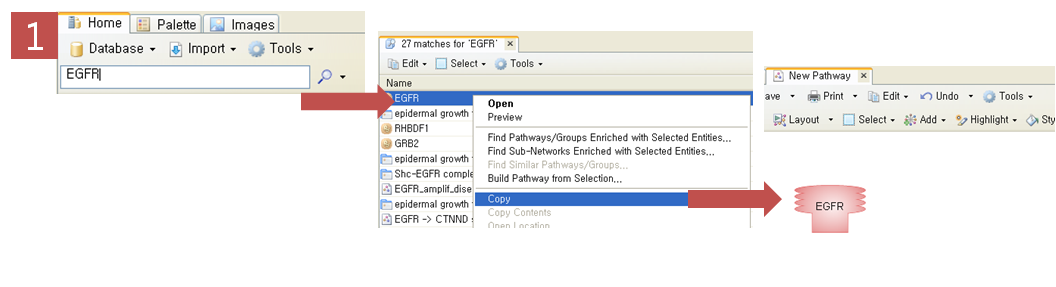 Step 2. Pathway 옵션 설정
Step 2. Pathway 옵션 설정
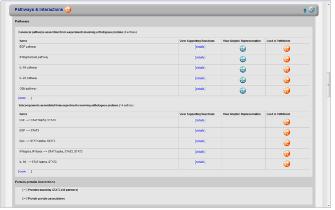
표피성장인자수용체(EGRF)에 의해 나타나는 질병을 모두 찾고 Pathway로 나타내기 위해 옵션 설정 과정을 거친다. Advanced
Build Pathway Wizard 에서 Add Neighbors > Directionality: “downstream”
> Entity type: “disease” > Filter Parameters: “regulation” 순으로
선택한다.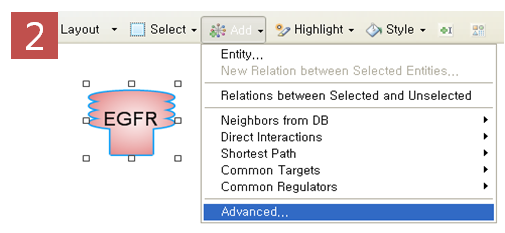 Step 3. Relation 속성을 이용한 검색 기능
Step 3. Relation 속성을 이용한 검색 기능
Pathway에서 “amplif”가 포함된
Regulation 관계만 찾기 위해 pathway 검색 tool에서 “Find Relation by Attribute”를
클릭한다. 찾고자 하는 Attribute에 대해 설정하는데 논문의 문장에 "amplif"가 포함된 것만 찾기위해
Attribute를 Sentence로 선택하고 Operation은 "includes", Value는 "amplif"로 설정하고
검색을 한다. 검색 결과 Reference sentence에 "amplif(amplification/amplified)"가 포함된
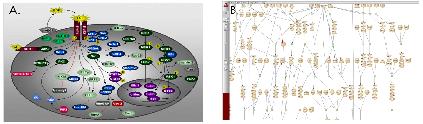
Relation만 파란색으로 표시된것을 확인할 수 있다.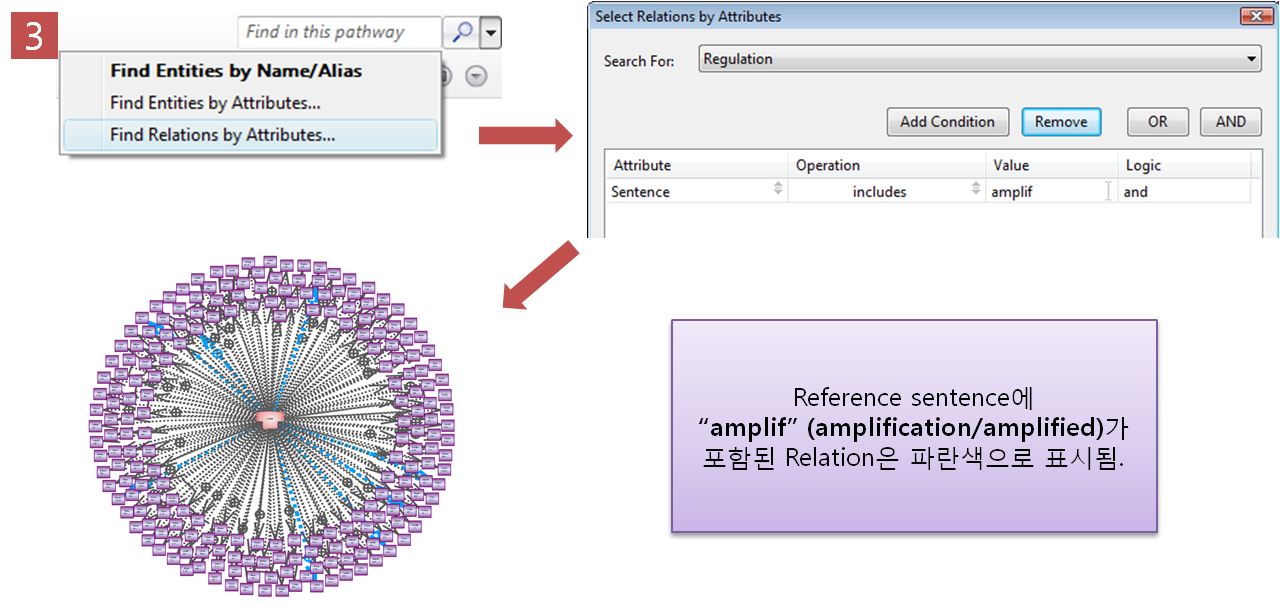 Step 4. 선택된 Pathway만 보기
Step 4. 선택된 Pathway만 보기
복잡한 Pathway에서 선택된 것만 자세히 보기 위해 Edit에서 Copy를 하고 새 Pathway 문서에 붙여넣기 한다. 그러면 EGFR이 증폭했을 때 생길 수 있는 질병에 대한 pathway만 확인 할 수 있다.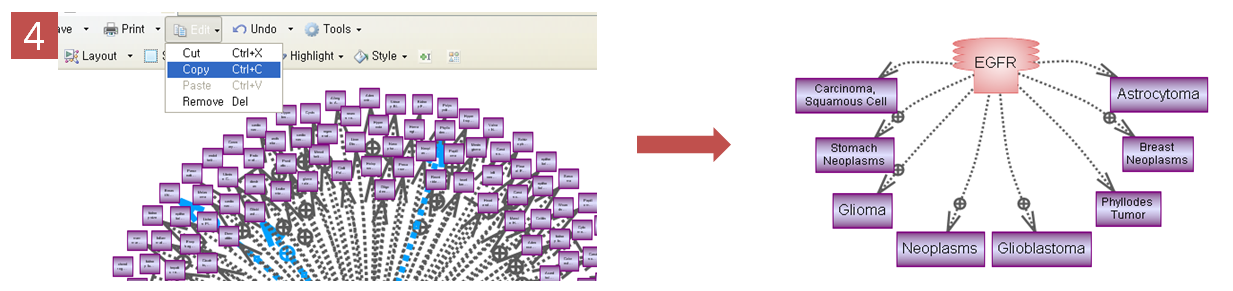
아래 동영상보기를 하시면 4개의 Step을 한 번에 보실 수 있습니다.
Posted by 人Co
- Tag
- EGFR, insilicogen, pathway, 인실리코젠, 증폭, 질병, 표피성장인자수용체
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/82