HGMD professional
- Posted at 2010/05/13 19:17
- Filed under 제품소식

HGMD는 현재 유전체 서열상의 변이로 인한 질병의 병변 및 유전자의 이름 그리고 유전체상의 위치 정보를 문헌정보에 근거하여 서비스하고 있다. 이러한 정보는 OMIM, Entrez Gene 그리고 Human Gene Nomenclature Committee를 포함한 대표 web-base 데이터들과 링크를 통해 변이에 의한 표현형, 구조적 정보들이 함께 제공되고 있다. 그 자세한 내용은 아래와 같다.
Feature
Up-to-date Mutation Data
- Full Coverage of PubMed journals
- Gene Centric Search
- Mutation Centric Search
- Reference Centric Search
- Boolean Full Text Searching
- View Mutation Data by Type
- View Mutation Data by Disease/Phenotype
- cDNA Sequences
- Extended cDNA Sequences
- Expanded Gene-specific Information
- Expanded Mutation-specific Information
- Advanced Search Tools
- Mutation Viewer/Maps
- Genomic Coordinates for Missense/Nonsense Mutations
- Search for Functional Polymorphisms
- HGVS Nomenclature for Missense/Nonsense Mutations
- Links to Entrez dbSNP (using rs numbers)
- Provision of Additional Literature References
- Search/Display of Gene Ontology Terms
- Downloadable Version
HGMD Professional은 위와 같이 변이 정보에 대한 서열 정보, SNP정보, HGVS nomenclature 정보를 링크를 통해 서비스하고 있으며 이들의 조절 메카니즘과 관련된 transcription factor 정보도 함께 지원하고 있다. 더욱이 이러한 모든 정보의 근간이 되는 문헌정보를 함께 제공하고 있어 그 신뢰성이 매우 높다 하겠다.
HGMD tutorial
Expanded Search Engine : 최신의 데이터를 사용자 편의에 의한 주제 중심의 인터페이스로 제공하고 있다. 키워드 방식을 이용한 특정 유전자, 질병의 상태, 변이정보, 문헌정보를 통한 검색이 가능하며 알파벳 인덱싱을 통한 검색도 가능하다.
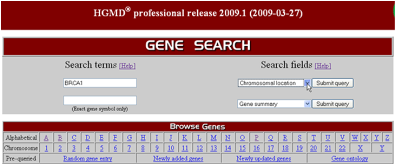
또한 특정 chromosome 내에 존재 하는 모든 변이정보를
한 번에 확인 할 수 도 있으며 이들 정보는 모두 다운 로드 기능을 통해 local PC에 저장이 가능하여
필요할 때 언제든지 활용이 가능 하다(Figure 2).
키워드 검색을 통한 유전자 검색의 경우 Figure 3에서 보여 지는 것과 같이 관련 유전자에서 동반 되는 모든 변이 정보를 확인할 수 있다. Splicing에 의한 변이정보, small deletion, small insertion 그리고 SNP에 의한 정보도 함께 검색 할 수 있다. 또한 transcription factor 정보도 링크되어 다양한 원인에 의해 유발되는 유전적 질환의 생화학적 정보를 통합적으로 확인 할 수 있다.
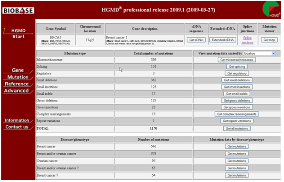
Figure 3. 유전자 검색. 유전자 검색을 통한 다양한 변이 정보 및 질병의 병변, 유전체상의 위치정보, 유전자 발현 조절 정보를 확인 할 수 있다.
Biochemical information : Human의 변이 정보는 구분된 카테고리 정보에서 keyword로 검색이 가능하며 이들의 정보는 이후 모두 다운로드가 가능하다. 질병의 phenotype을 비롯한 유전체 상의 위치 정보, dbSNP와 같은 기존 참조 데이터베이스의 정보, motif, regulation, 참조 문헌 정보까지 한 번에 확인할 수 있다.
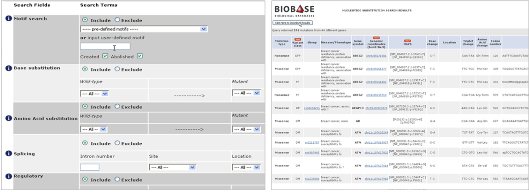
Figure 4. 변이 정보 검색 결과. 유전적 변이에 의한 DNA서열의 변화, 단백질 서열 변이, 참조 데이터베이스, phenotype, gene ontology, 참조 문헌 정보를 모두 다운 받을 수 있다.
Personal genomics 시대에 가장 필수적인 데이터베이스중 하나인 Biobase HGMD는 개인의 잠재적인 유전적 질환의 탐색부터 현재 발병중인 질환에 대한 치료 연구를 위해 많은 기초 데이터를 제공할 것으로 여겨진다. 많은 논문과 데이터베이스를 집대성하여 유전적 질환의 통합적 정보를 제공하고 있는 HGMD는 앞으로 더 많은 연구자들에게 도움이 될 것이다.

Posted by 人Co
- Tag
- BIOBASE, dbSNP, deletion, Entrez Gene, HGMD, HGVS nomenclature, Human Gene Nomenclature Committee, insertion, insilicogen, motif, mutation, NGS, OMIM, personal genomics, phenotype, regulation, SNP, splicing, 돌연변이, 인실리코젠, 질병
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/73