식물 리그닌화(목질화)에 대한 후보 유전자 리스트 확인하기
- Posted at 2010/12/27 09:01
- Filed under 제품소식
How can I find literature supporting a list of candidate genes related to a specific plant trait?
식물 리그닌화(목질화)에 대한 후보 유전자 리스트를 확인할 수 있는 있는가?
리그닌은 목질소라고도 하며 셀룰로오스, 헤미셀룰로오스와 더불어 식물골격 구성성분의 하나로 목재 20~30%에 달한다. 식물의 리그닌화(목질화)는 식물이 자라는 과정에서 세포막이나 중간층에 리그닌이 생겨 흡착되거나 화학적으로 결합하여 강하고 단단해지는 현상을 말한다. 일반적으로 세포의 성장에 따라 서서히 일어나지만 병태 또는 상해 조직에서는 유조직세포벽에 급속히 일어나는 경우도 있다. 목질화된 세포벽은 병원균에 의해 잘 침범되지 않기 때문에 병태조직 또는 상해조직에서의 목질화는 일종의 방어반응이라고 할 수 있다.
Step to follow
Step 1. Gene list 검색
검색하고자 하는 유전자의 TAIR ID를 검색한다. Import > Gene List > 복사해 놓은 TAIR ID 리스트 Paste from Clipboard > Lookup in the Database 클릭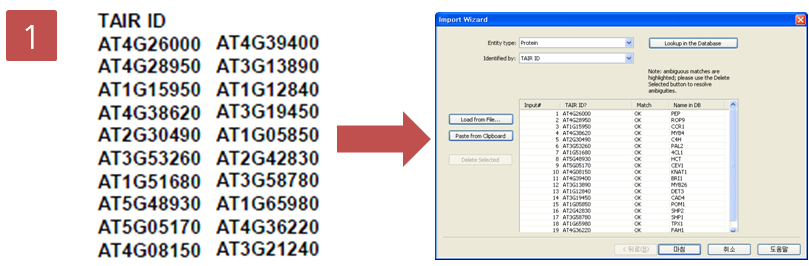
Step 2. Lignification과 관련있는 Cell Process 검색
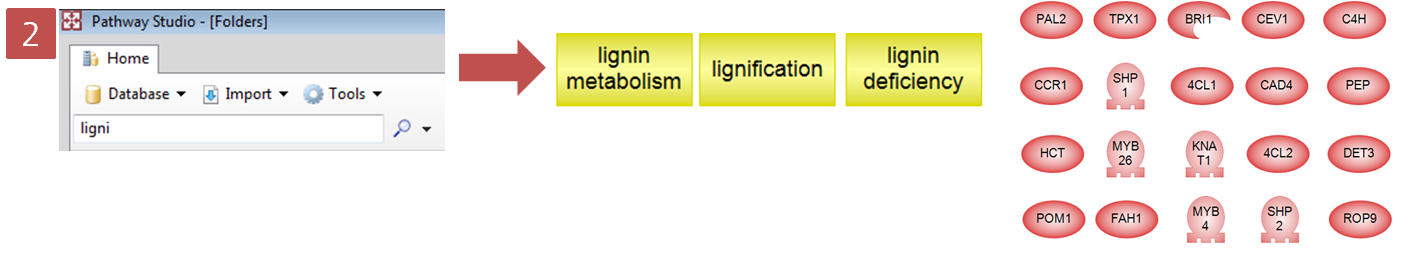 Step 3. Pathway 옵션 설정
Step 3. Pathway 옵션 설정
Protein과 Cell Process의 Relation 사이의 관계를 나타내기 위해 Protein을 선택한 상태에서 Add 메뉴를 선택하고 "Relations between Selected and Unselected" 클릭 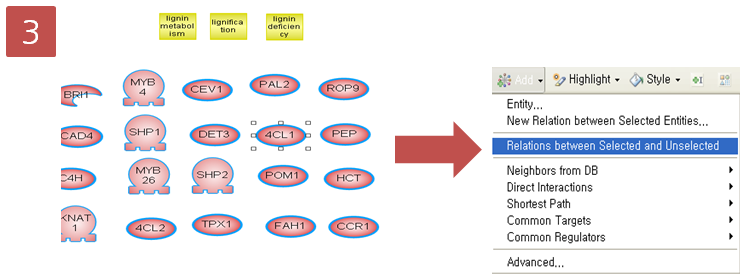 Step 4. Pathway 확인
Step 4. Pathway 확인
"Relations between Selected and Unselected"를 하면 서로 연관이 있는 Relation만 표시가 되고 연관이 없는 Entity는 Relation이 되지 않고 남아있다.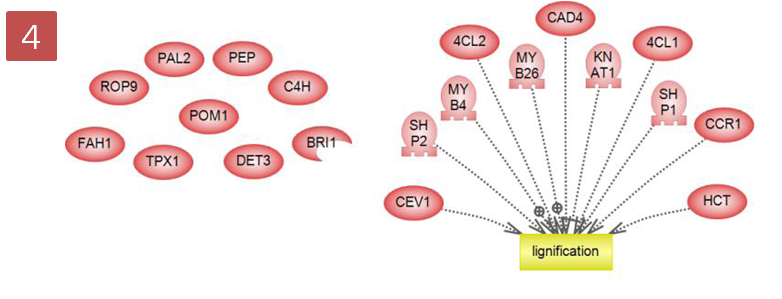 Step 5. 엔티티 추가하기
Step 5. 엔티티 추가하기
"lignin" small molecule을 검색하여 4번 pathway에 추가하고 Protein과 "lignin" 사이의 Relation을 확인하기 위해 "lingin"을 선택한 다음 Step 3을 반복한다.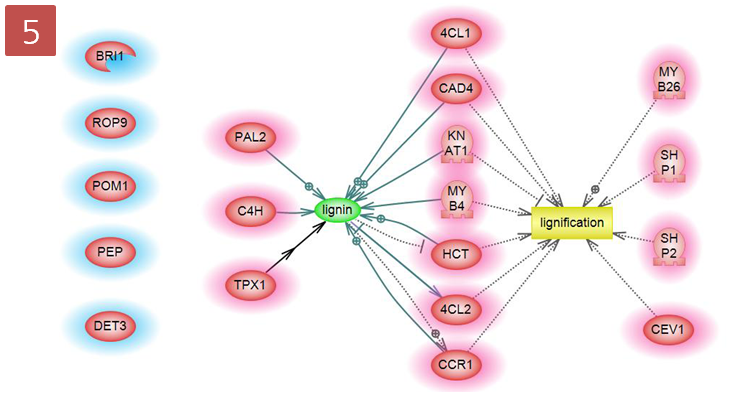
Posted by 人Co
- Tag
- Ariadne, insilicogen, lignification, lignin, pathway, PathwayStudio, 리그닌, 목질화, 식물 리그닌화, 인실리코젠
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/88