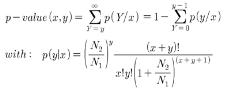NGS 분석전략 세미나 개최 후기
- Posted at 2010/02/25 17:37
- Filed under 회사소식
지난 2월 5일, 저희 (주)인실리코젠의 Codes팀은 "Practical bioinformatics pipeline for NGS data"라는 주제로 세미나를 개최하였습니다.
 이번 교육은 당사에서 발간한 Quipu Issue Paper 2호의 "NGS 시대의 분석전략 2"을 중심으로 최근 가장 이슈가
되고 있는 NGS 데이터의 assembly, 그리고 그 이후에 진행할 수 있는 다양한 분석들에 대한 내용들을 크게 3가지
세션으로 나누어 구성하였습니다. 또한 생물정보 분야의 중심 역할을 하고 있는 한국생명공학연구원
국가생물자원정보관리센터(KOBIC)의 많은 연구원분들을 대상으로 진행되었습니다.
이번 교육은 당사에서 발간한 Quipu Issue Paper 2호의 "NGS 시대의 분석전략 2"을 중심으로 최근 가장 이슈가
되고 있는 NGS 데이터의 assembly, 그리고 그 이후에 진행할 수 있는 다양한 분석들에 대한 내용들을 크게 3가지
세션으로 나누어 구성하였습니다. 또한 생물정보 분야의 중심 역할을 하고 있는 한국생명공학연구원
국가생물자원정보관리센터(KOBIC)의 많은 연구원분들을 대상으로 진행되었습니다.
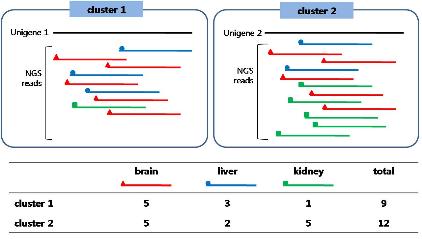
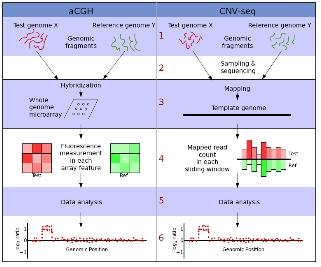
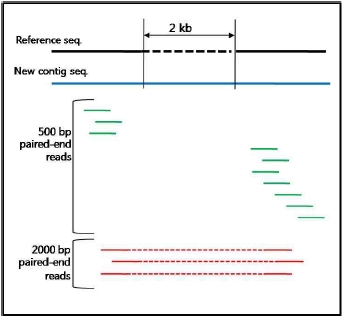
 NGS 데이터의 assembly는 유전체 분석에 있어서 데이터 플랫폼의 종류와 어떤 어셈블러를 사용하느냐에 따른 분석 전략 및
파이프라인은 꼭 필요할 것이라 생각합니다. 이에 첫 번째 세션은 De novo assembly와 Reference
assembly에 사용되고 있는 여러 가지 어셈블러들의 종류, 장단점 비교, 실제 데이터 벤치마킹 결과 등에 대한 내용으로
준비하였고, 발표 중간중간 관련 사항에 대한 질문과 열띤 토론으로 참석하신 연구원분들의 많은 관심을 받았습니다.
NGS 데이터의 assembly는 유전체 분석에 있어서 데이터 플랫폼의 종류와 어떤 어셈블러를 사용하느냐에 따른 분석 전략 및
파이프라인은 꼭 필요할 것이라 생각합니다. 이에 첫 번째 세션은 De novo assembly와 Reference
assembly에 사용되고 있는 여러 가지 어셈블러들의 종류, 장단점 비교, 실제 데이터 벤치마킹 결과 등에 대한 내용으로
준비하였고, 발표 중간중간 관련 사항에 대한 질문과 열띤 토론으로 참석하신 연구원분들의 많은 관심을 받았습니다.
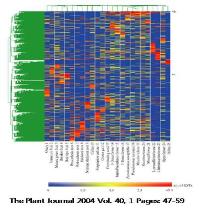
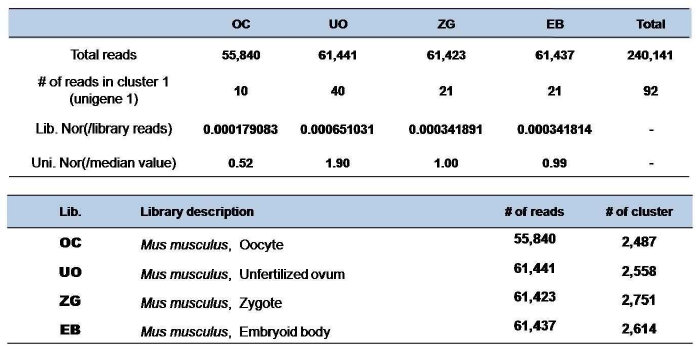
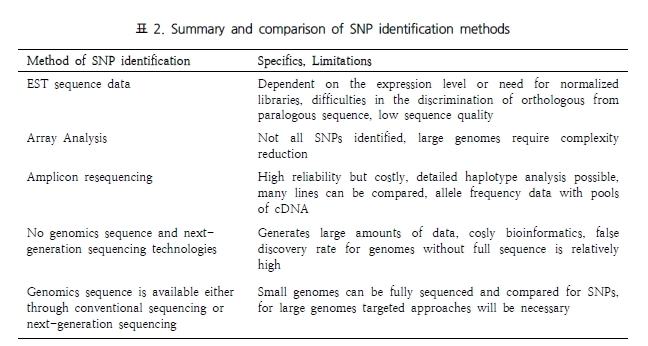
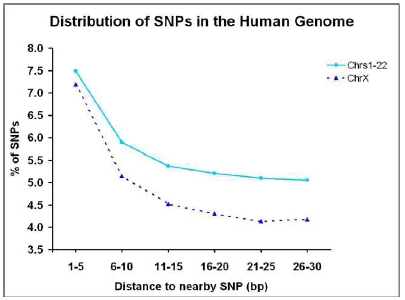
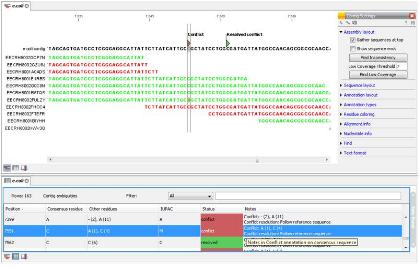
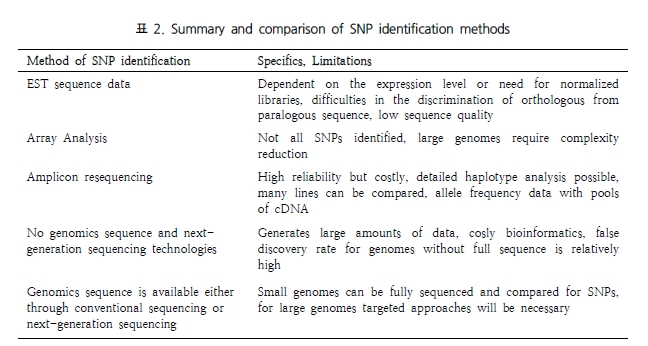
 두번째 세션은 SNP 분석 방법 및 최근 capture array 분석의 실제 연구사례, 관련 솔루션 등을 소개한
variation 분석 파트와 EST 데이터를 이용한 functional annotation, Organism-specific
분석, Ortholog/Paralog 유전자 분석방법 등에 대한 expression 분석 파트로 구분되어 진행되었으며 마지막
세션은 NGS와 생물정보 파이프라인을 이용한 Genome annotation에 대한 내용으로 현재 NGS 염기서열 결정 이후
문제점 및 이슈를 분석하고 효율적인 전략들을 소개하였습니다. 또한 structural annotation과 functional
annotation의 분석 방법 및 실제 Codes팀의 분석 컨설팅 파이프라인 관련하여도 설명 드릴 수 있는 좋은시간이
되었습니다.
두번째 세션은 SNP 분석 방법 및 최근 capture array 분석의 실제 연구사례, 관련 솔루션 등을 소개한
variation 분석 파트와 EST 데이터를 이용한 functional annotation, Organism-specific
분석, Ortholog/Paralog 유전자 분석방법 등에 대한 expression 분석 파트로 구분되어 진행되었으며 마지막
세션은 NGS와 생물정보 파이프라인을 이용한 Genome annotation에 대한 내용으로 현재 NGS 염기서열 결정 이후
문제점 및 이슈를 분석하고 효율적인 전략들을 소개하였습니다. 또한 structural annotation과 functional
annotation의 분석 방법 및 실제 Codes팀의 분석 컨설팅 파이프라인 관련하여도 설명 드릴 수 있는 좋은시간이
되었습니다.
 이렇게 바쁜 와중에도 하루의 일정을 직접 방문하여 소화해주신 KOBIC 연구원분들께 감사의 인사를 드리며, 진행된 교육으로
인해서 NGS 데이터를 분석하고 연구하시는데 조금이나마 도움이 되었으면 하는 바램입니다. 또한 "NGS시대의 분석전략 3"의
발간도 부탁하실 정도로 기술소식지와 세미나에 큰 관심을 보여주셔서 더욱 뜻 깊은 시간이었고, 앞으로도 이러한 교육의 자리를 많이
준비하도록 노력하겠습니다.
이렇게 바쁜 와중에도 하루의 일정을 직접 방문하여 소화해주신 KOBIC 연구원분들께 감사의 인사를 드리며, 진행된 교육으로
인해서 NGS 데이터를 분석하고 연구하시는데 조금이나마 도움이 되었으면 하는 바램입니다. 또한 "NGS시대의 분석전략 3"의
발간도 부탁하실 정도로 기술소식지와 세미나에 큰 관심을 보여주셔서 더욱 뜻 깊은 시간이었고, 앞으로도 이러한 교육의 자리를 많이
준비하도록 노력하겠습니다.
 책자로 발간되었지만, 이번 세미나 내용을 포함한 NGS시대의 분석전략은 더욱 많은 연구자분들께 유익한 정보를 제공해 드리고자 블로그 연재도 계속 진행중입니다. 이와 관련한 자세한 문의사항은 저희 (주)인실리코젠의 Codes팀에게 연락 부탁드립니다.
책자로 발간되었지만, 이번 세미나 내용을 포함한 NGS시대의 분석전략은 더욱 많은 연구자분들께 유익한 정보를 제공해 드리고자 블로그 연재도 계속 진행중입니다. 이와 관련한 자세한 문의사항은 저희 (주)인실리코젠의 Codes팀에게 연락 부탁드립니다.
(Tel: 031-278-0061, E-mail: codes@insilicogen.com)
(Tel: 031-278-0061, E-mail: codes@insilicogen.com)
Posted by 人Co
- Tag
- annotation, Assembly, Bioinformatics, capture array, de novo assembly, Expression, insilicogen, KOBIC, NGS, Reference assembly, SNP
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/48