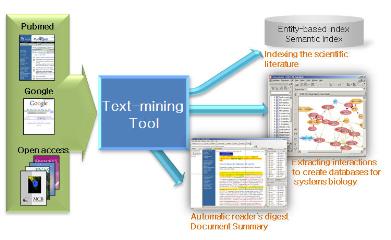
[Quipu Issue Paper] Bioinformatics Knowledge Management Ⅴ- Centralization for High-throughput Data Analysis
- Posted at 2010/04/09 09:42
- Filed under 생물정보
연재 순서
1. Assembly
2. Variation study
3. Expression study
4. Epigenomics
5. Genome Annotation
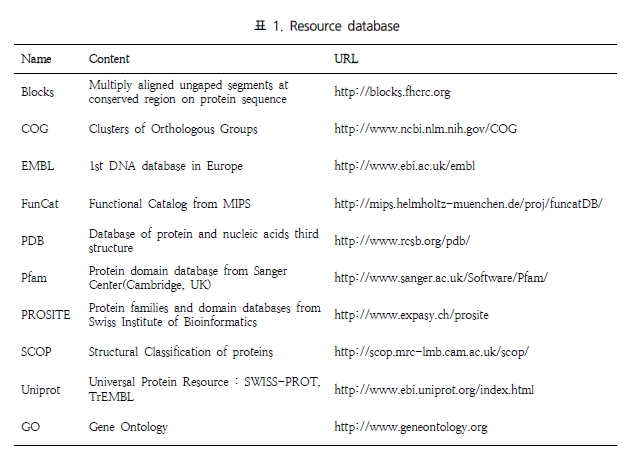
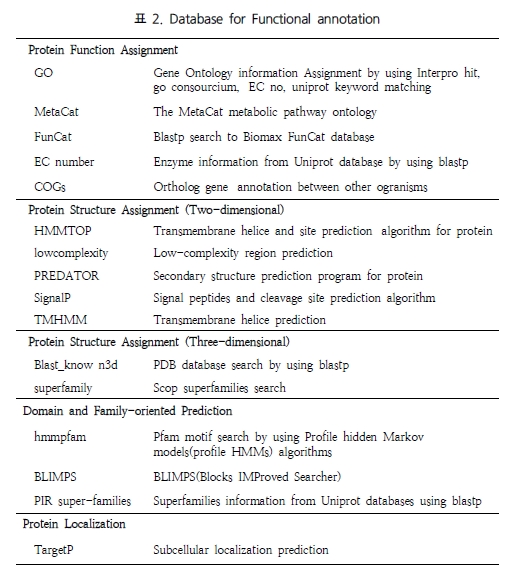
6. Next Generation Bioinformatics
7. Data Management for web 2.0 Era
8. Semantic Network for Integrated Biology Data
9. Gene Network Discovery by Text-mining
10. Centralization for High-throughput Data Analysis
이번 연재에서는 NGS Edition의 마지막 연재로 대용량의 데이터를 다루기 위한 Centralization for High-throughput Data Analysis에 대해 알아보도록 하겠습니다.
최근 들어 분석하고자 하는 데이터의 용량이 기하급수적으로 늘어남에 따라 데스크탑 컴퓨터 사양으로 분석하기가 어려워지고 있다. 따라서 생물정보 전문가들의 도움이 많이 요구되지만, 한 두 명의 생물정보 전문가들이 처리하기에는 분석하고자 하는 데이터가 급격하게 증가되고 이를 활용한 연구 분야가 다양하여 대규모의 생물정보 전문가를 가용하고 있는 센터가 아닌 곳에서 모든 분석을 지원하는 것은 쉽지가 않다. 또한 유전체 분석과 같은 대규모 프로젝트가 컨소시엄 형식으로 수행되고 있는 상황에서는 다른 연구팀과의 상호 협조를 통한 공동 연구가 중요하며, 이를 위한 데이터의 공유와 관리도 중요시되고 있다. 따라서 연구자들이 공동으로 데이터를 업데이트하거나 다운로드할 수 있는 데이터베이스와 대규모의 용량을 분석할 수 있는 서버, 그리고 서버에서 분석한 결과를 개별 컴퓨터에서 확인할 수 있는 시스템의 유기적인 관계가 요구된다. 하지만 생물데이터의 형식과 이를 분석하는 프로그램의 종류가 다양하므로 데이터의 공유와 관리, 그리고 분석 프로그램의 연계가 상당히 복잡하다.
대다수의 생물학자들이 윈도우 운영체제의 컴퓨터를 사용하고 있으며 Vector NTI, DNA Star와 같은 생물데이터를 분석하는 상용화 프로그램을 많이 이용하고 있다. 하지만 이런 상용화 소프트웨어는 윈도우에서만 사용가능하며, 분석하는 데이터의 용량 및 길이에 제한을 두고있으므로, 대규모의 데이터를 분석하는 것은 적절하지 않다.
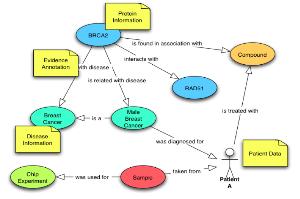
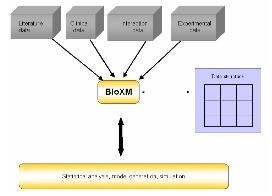
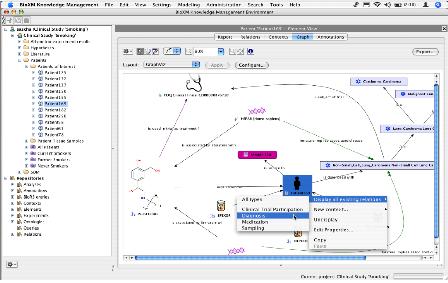
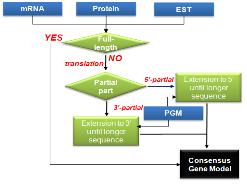
CLC bio사에서는 대규모의 NGS 데이터 및 대규모의 데이터를 서버에서 분석할 수 있는 CLC Genomics Server(그림 11)와 데스크탑 컴퓨터에서 결과를 확인하고 Vector NTI, DNA Star와 같은 다양한 분석 프로그램에서 나온 결과 데이터를 사용할 수 있는 CLC Genomics Workbench를 개발하였다.
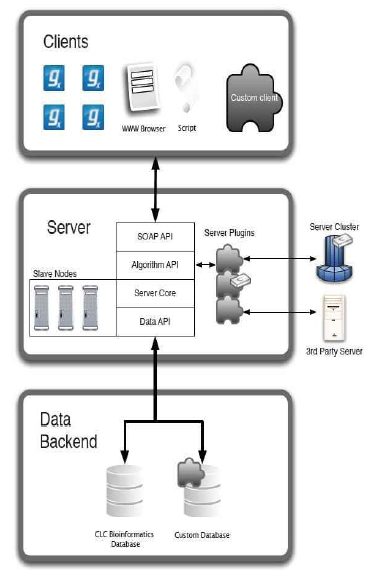 그림 11. Genomics Server 시스템 아키텍처
그림 11. Genomics Server 시스템 아키텍처
3-5. Centralization for High-throughput Data Analysis
최근 들어 분석하고자 하는 데이터의 용량이 기하급수적으로 늘어남에 따라 데스크탑 컴퓨터 사양으로 분석하기가 어려워지고 있다. 따라서 생물정보 전문가들의 도움이 많이 요구되지만, 한 두 명의 생물정보 전문가들이 처리하기에는 분석하고자 하는 데이터가 급격하게 증가되고 이를 활용한 연구 분야가 다양하여 대규모의 생물정보 전문가를 가용하고 있는 센터가 아닌 곳에서 모든 분석을 지원하는 것은 쉽지가 않다. 또한 유전체 분석과 같은 대규모 프로젝트가 컨소시엄 형식으로 수행되고 있는 상황에서는 다른 연구팀과의 상호 협조를 통한 공동 연구가 중요하며, 이를 위한 데이터의 공유와 관리도 중요시되고 있다. 따라서 연구자들이 공동으로 데이터를 업데이트하거나 다운로드할 수 있는 데이터베이스와 대규모의 용량을 분석할 수 있는 서버, 그리고 서버에서 분석한 결과를 개별 컴퓨터에서 확인할 수 있는 시스템의 유기적인 관계가 요구된다. 하지만 생물데이터의 형식과 이를 분석하는 프로그램의 종류가 다양하므로 데이터의 공유와 관리, 그리고 분석 프로그램의 연계가 상당히 복잡하다.
대다수의 생물학자들이 윈도우 운영체제의 컴퓨터를 사용하고 있으며 Vector NTI, DNA Star와 같은 생물데이터를 분석하는 상용화 프로그램을 많이 이용하고 있다. 하지만 이런 상용화 소프트웨어는 윈도우에서만 사용가능하며, 분석하는 데이터의 용량 및 길이에 제한을 두고있으므로, 대규모의 데이터를 분석하는 것은 적절하지 않다.
CLC bio사에서는 대규모의 NGS 데이터 및 대규모의 데이터를 서버에서 분석할 수 있는 CLC Genomics Server(그림 11)와 데스크탑 컴퓨터에서 결과를 확인하고 Vector NTI, DNA Star와 같은 다양한 분석 프로그램에서 나온 결과 데이터를 사용할 수 있는 CLC Genomics Workbench를 개발하였다.
CLC Genomics Workbench에서 CLC Genomics Server에 NGS 데이터 및 대규모 분석 데이터를 업데이트하고 분석을 수행한 뒤 CLC Genomic Server에서 분석되어진 결과를 CLC Genomics Workbench에서 확인할 수 있는 플러그인이 있다. 이를 활용하면 대규모 리소스를 필요로 하는 데이터의 분석과 데스크탑 컴퓨터에서 가능한 데이터 분석을 구분하여 연구 업무의 효율성을 증대시킬 수 있다. 또한 윈도우, 리눅스, 매킨토시 등 운영체제에 관계없이 설치가 가능하기 때문에 다양한 운영체제에서 데이터를 분석하는 연구자들이 분석결과를 공유할 수 있다. 대부분의 상용화 프로그램은 연구자들이 원하는 분석 알고리즘이 없을 경우 이후 버전의 업그레이드 내용을 기다리거나, 다른 프로그램을 이용하여 분석할 수밖에 없으므로 분석의 일관성을 유지하기 어렵고, 번거로움이 가증되었다.
하지만, CLC Genomics Server에서는 External Application 플러그인을 적용하여 CLC Genomics Workbench에 설치되어 있지 않는 알고리즘 및 분석법을 커맨드라인 방식으로 설치한 후 간단한 설정을 통해 별도의 인터페이스를 만들지 않더라도 CLC Genomics Workbench에서 데이터의 입력과 출력을 수행할 수 있으며, 분석 결과를 다른 분석에 응용할 수 있다.
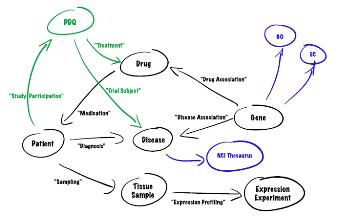
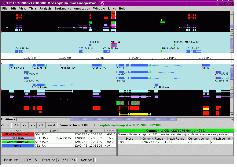
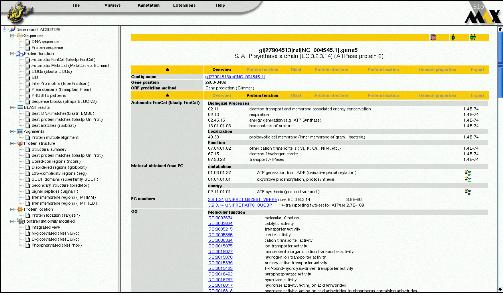
그림 12는 CLC Genomics Server에서 external application 모듈을 설정하는 것을 보이고 있으며, 그림 13은 external application을 통해서 구축한 새로운 모듈을 이용하여 분석하는 화면을 보이고 있다. 이와 같이 서버급에서 분석할 수 있는 시스템과 데스크탑 컴퓨터에서 분석할 수 있는 프로그램의 연계를 통해서 생물학자들이 복잡하고 다양한 데이터를 분석하는데 많은 도움을 줄 수 있을 것이다.
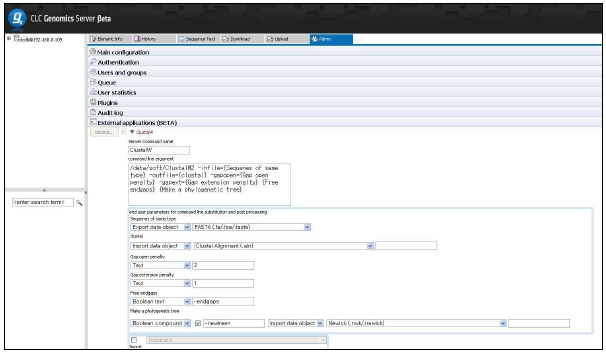 그림 12. External Application of CLC Genomics Server. 자주 사용되는 커맨드라인 방식의 프로그램은 CLC Genomics Server의 External Application 설정을 통해 별도의 인터페이스를 만들지 않고 CLC Genomics Workbench에서 수행할 수 있다. 이를 이용하여 사용자에 맞춰진 workbench로 재구성할 수 있다.
그림 12. External Application of CLC Genomics Server. 자주 사용되는 커맨드라인 방식의 프로그램은 CLC Genomics Server의 External Application 설정을 통해 별도의 인터페이스를 만들지 않고 CLC Genomics Workbench에서 수행할 수 있다. 이를 이용하여 사용자에 맞춰진 workbench로 재구성할 수 있다.
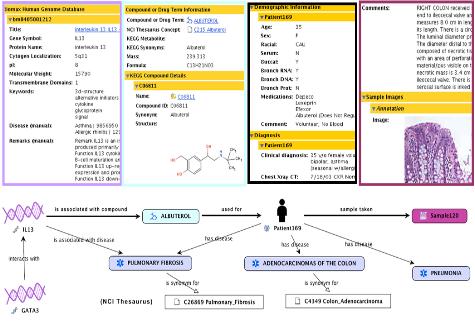
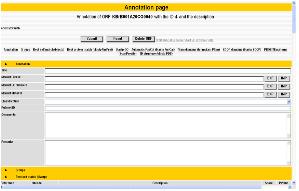
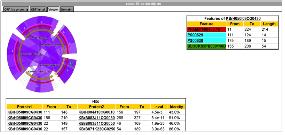
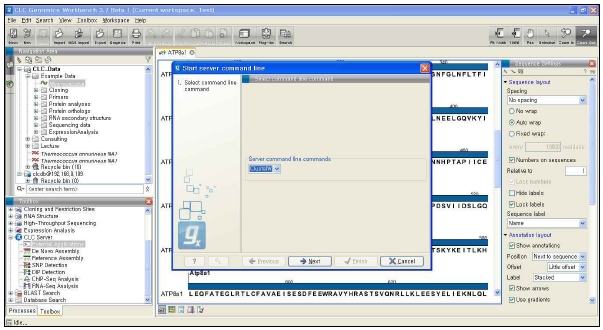 그림 13. CLC Genomics Workbench 플러그인 적용. External application 플러그인으로 구축된 새로운 모듈은 CLC Genomics Workbench에서 분석이 가능하다.
그림 13. CLC Genomics Workbench 플러그인 적용. External application 플러그인으로 구축된 새로운 모듈은 CLC Genomics Workbench에서 분석이 가능하다.
저희 (주)인실리코젠 Codes팀은 최신 생물정보학관련 연구 동향에 대한 기술 소식지(Quipu Issue Paper)를 발간하고 이 소식지를 통하여 빠르게 발전하는 NGS 시대에 다양한 변화를 습득하고 하시는 연구에 조금이나마 도움이 되길 바라면서 지난 2월부터 약 2개월에 걸쳐 저희 회사 블로그 Quipu(http://www.insilicogen.com/blog/)를 통해 연재를 진행하였습니다. 지난 2개월 동안 최신 생물정보학관련 연구 동향에 대한 기술 소식지 블로그 연재에 많은 관심 가져주셔서 진심으로 감사드리며 저희 (주)인실리코젠은 앞으로도 생물정보 분야에서 끊임없이 노력하는 기업이 되겠습니다.
(주)인실리코젠 Codes팀 배상
Tel: 031-278-0061 / E-mail: codes@insilicogen.com
하지만, CLC Genomics Server에서는 External Application 플러그인을 적용하여 CLC Genomics Workbench에 설치되어 있지 않는 알고리즘 및 분석법을 커맨드라인 방식으로 설치한 후 간단한 설정을 통해 별도의 인터페이스를 만들지 않더라도 CLC Genomics Workbench에서 데이터의 입력과 출력을 수행할 수 있으며, 분석 결과를 다른 분석에 응용할 수 있다.
그림 12는 CLC Genomics Server에서 external application 모듈을 설정하는 것을 보이고 있으며, 그림 13은 external application을 통해서 구축한 새로운 모듈을 이용하여 분석하는 화면을 보이고 있다. 이와 같이 서버급에서 분석할 수 있는 시스템과 데스크탑 컴퓨터에서 분석할 수 있는 프로그램의 연계를 통해서 생물학자들이 복잡하고 다양한 데이터를 분석하는데 많은 도움을 줄 수 있을 것이다.
저희 (주)인실리코젠 Codes팀은 최신 생물정보학관련 연구 동향에 대한 기술 소식지(Quipu Issue Paper)를 발간하고 이 소식지를 통하여 빠르게 발전하는 NGS 시대에 다양한 변화를 습득하고 하시는 연구에 조금이나마 도움이 되길 바라면서 지난 2월부터 약 2개월에 걸쳐 저희 회사 블로그 Quipu(http://www.insilicogen.com/blog/)를 통해 연재를 진행하였습니다. 지난 2개월 동안 최신 생물정보학관련 연구 동향에 대한 기술 소식지 블로그 연재에 많은 관심 가져주셔서 진심으로 감사드리며 저희 (주)인실리코젠은 앞으로도 생물정보 분야에서 끊임없이 노력하는 기업이 되겠습니다.
(주)인실리코젠 Codes팀 배상
Tel: 031-278-0061 / E-mail: codes@insilicogen.com
Posted by 人Co
- Tag
- CLC bio, CLC Genomics Serer, CLC Genomics Workbench, Codes, DNA Star, External Application, High-throughput, insilicogen, NGS, quipu, VectorNTI, 인실리코젠 블로그
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/68