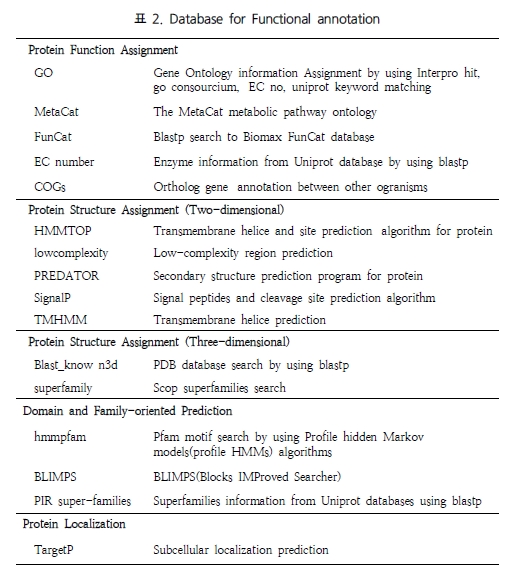
[Quipu Issue Paper] Bioinformatics Knowledge Management Ⅱ- Data Management for Web 2.0 Era
- Posted at 2010/03/30 14:53
- Filed under 생물정보
연재 순서
1. Assembly
2. Variation study
3. Expression study
4. Epigenomics
5. Genome Annotation
6. Next Generation Bioinformatics
7. Data Management for web 2.0 Era
8. Semantic Network for Integrated Biology Data
9. Gene Network Discovery by Text-mining
10. Centralization for High-throughput Data Analysis
이번 연재에서는 웹 2.0 시대에 맞추어 다양한 분야에서 생산된 데이터를 효율적으로 관리하는 방법에 대해 알아보겠습니다.
3-2. Data Management for Web 2.0 Era
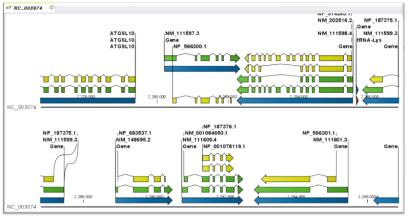
최근 들어 웹 2.0에 대한 논란이 뜨겁다. 비단 IT에서 뿐만 아니라 일상생활에서도 웹 2.0이라는 용어는 어렵지 않게 들을 수 있게 되었다. ‘O'Reilly Media’에서 2003년 처음 사용하기 시작하면서부터 대중화 된 웹 2.0에 대한 정의를 위키피디아에서는 이렇게 정의하고 있다. 단순한 웹사이트의 집합체를 웹 1.0으로 보고 있다면, 웹 2.0은 웹 애플리케이션을 제공하는 하나의 완전한 플랫폼으로의 발전이다(그림 1)[2].

웹 2.0이 세상에 알려진 지 불과 몇 년 밖에 되지 않았으나, 이제는 누구나가 쉽게 웹 2.0 으로 구축된 사이트에 자신의 데이터를 올리거나 다른 사람의 데이터를 다운로드 받는 등 이전보다 능동적으로 웹을 활용하고 있다. 위키라는 개념으로 접근한 웹 2.0은 대영백과사전으로 알려져 있는 브리태니커 백과사전보다 더 많은 정보를 담고 있어 세계 최대의 백과사전으로 기네스북에 기록되어 있다. 2001년 1월 출범하여 2006년 12월에 기네스북에 인정되기까지 불과 6년의 짧은 기간에 엄청난 정보가 모이게 된 가장 큰 원동력은 웹 2.0 시대에 맞춰 운영자가 절대 개입하지 않는다는 원칙하에 모든 방문자가 스스로 글을 읽고 쓸 수 있게 한 것이다. 정보의 양을 기준으로 한다면 다양한 사람들이 생산하는 데이터가 한 명이 작성한 글보다 훨씬 클 수 있으며, 다양한 사람들에 의해서 읽혀지고 수정되어 보다 정확한 정보들로 탈바꿈할 수 있다는 것이다.
(주)인실리코젠에서는 웹 2.0 시대에 맞추어 위키 시스템을 활용한 홈페이지 형식의 지식관리시스템을 재구성하였다. 이를 이용하여 프로젝트 단위의 연구에서 공동 연구자들 사이의 데이터 공유 또는 미팅과 관련된 로그 관리 등을 효율적으로 운영하고 있다. 개별 연구자들이 각자 분석업무를 업로드하고 관련 연구자들이 업데이트된 데이터를 다운로드 받거나 웹상에서 오류나 문제점을 수정, 지적하여 최종적으로는 다양한 연구 전문가 그룹에 의한 완성된 연구 결과를 도출하는 방식을 취하고 있다. 이와 같은 방식의 연구 관리는 데이터가 실험실에 존재하는 것이 아니라 웹상에 존재하면서, 인터넷만 연결된 환경에서는 장소, 시간에 관계없이 공통의 주제를 가지고 분석 업무를 수행하고 데이터를 보완 할 수 있다는 장점이 있다. 한 사람의 불완전한 지식에서 시작하여 공동 연구자 또는 다른 전문가의 지식을 포함하여 완전한 지식을 유도하는 방향을 추구하고 있는 것이다.
이처럼 다양한 분야의 생물학 영역에서도 웹 2.0의 장점을 백분 발휘하여 데이터를 축적하고, 보완하고, 관리하는 흐름을 엿볼 수 있다. 최근에는 웹 2.0의 장점에 덧붙여서 다양한 분야에서 생산된 데이터를 어떻게 의미론적으로 재분류할 수 있는가에 보다 많은 관심이 집중되고 있다. 기존에 존재하는 지식 자원을 연구자가 검색, 통합 및 관리하기 편리하도록 의미기반 온톨로지 사이트로 구축하는 것이다. 그리하여, 최근의 많은 포탈검색사이트에서 입력된 검색어에 관한 다양한 관련 정보를 추출하여 보여주는 의미론적 검색 방법을 개발하였으며, 한국인들이 가장 많이 검색하는 네이버에서도 의미검색사이트를 개발하여 서비스하고 있다.
다음 연재에서는 생물학의 많은 데이터를 활용하여 새로운 의미를 발굴할 수 있는 Semantic Network for Integrated Biology Data에 대해 알아보겠습니다.
많은 관심 부탁드립니다.
참고문헌
(주)인실리코젠에서는 웹 2.0 시대에 맞추어 위키 시스템을 활용한 홈페이지 형식의 지식관리시스템을 재구성하였다. 이를 이용하여 프로젝트 단위의 연구에서 공동 연구자들 사이의 데이터 공유 또는 미팅과 관련된 로그 관리 등을 효율적으로 운영하고 있다. 개별 연구자들이 각자 분석업무를 업로드하고 관련 연구자들이 업데이트된 데이터를 다운로드 받거나 웹상에서 오류나 문제점을 수정, 지적하여 최종적으로는 다양한 연구 전문가 그룹에 의한 완성된 연구 결과를 도출하는 방식을 취하고 있다. 이와 같은 방식의 연구 관리는 데이터가 실험실에 존재하는 것이 아니라 웹상에 존재하면서, 인터넷만 연결된 환경에서는 장소, 시간에 관계없이 공통의 주제를 가지고 분석 업무를 수행하고 데이터를 보완 할 수 있다는 장점이 있다. 한 사람의 불완전한 지식에서 시작하여 공동 연구자 또는 다른 전문가의 지식을 포함하여 완전한 지식을 유도하는 방향을 추구하고 있는 것이다.
이처럼 다양한 분야의 생물학 영역에서도 웹 2.0의 장점을 백분 발휘하여 데이터를 축적하고, 보완하고, 관리하는 흐름을 엿볼 수 있다. 최근에는 웹 2.0의 장점에 덧붙여서 다양한 분야에서 생산된 데이터를 어떻게 의미론적으로 재분류할 수 있는가에 보다 많은 관심이 집중되고 있다. 기존에 존재하는 지식 자원을 연구자가 검색, 통합 및 관리하기 편리하도록 의미기반 온톨로지 사이트로 구축하는 것이다. 그리하여, 최근의 많은 포탈검색사이트에서 입력된 검색어에 관한 다양한 관련 정보를 추출하여 보여주는 의미론적 검색 방법을 개발하였으며, 한국인들이 가장 많이 검색하는 네이버에서도 의미검색사이트를 개발하여 서비스하고 있다.
다음 연재에서는 생물학의 많은 데이터를 활용하여 새로운 의미를 발굴할 수 있는 Semantic Network for Integrated Biology Data에 대해 알아보겠습니다.
많은 관심 부탁드립니다.
참고문헌
2. http://ko.wikipedia.org/wiki/Web2.0
Posted by 人Co
- Tag
- Bioinformatics, NGS, 웹 2.0, 위키, 위키피디아, 인실리코젠
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/65