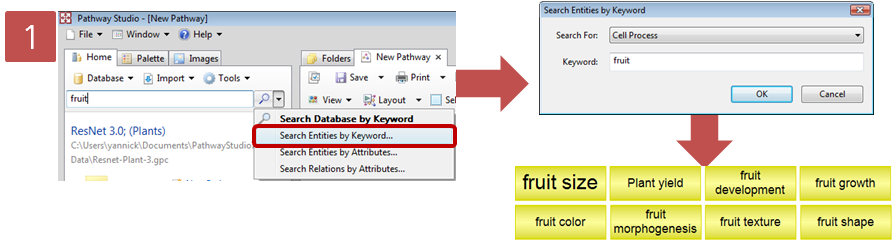
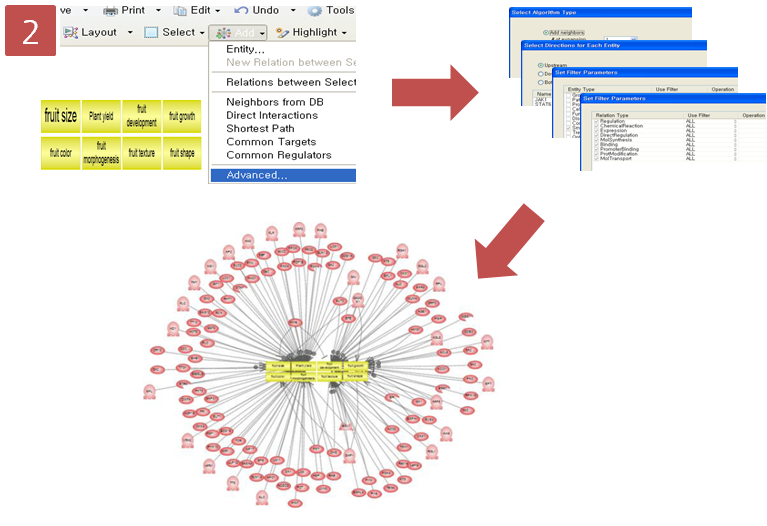
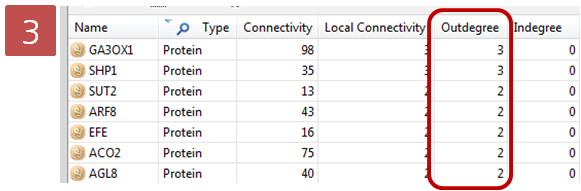
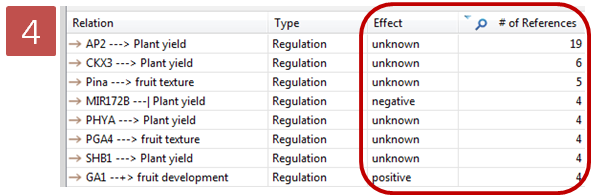
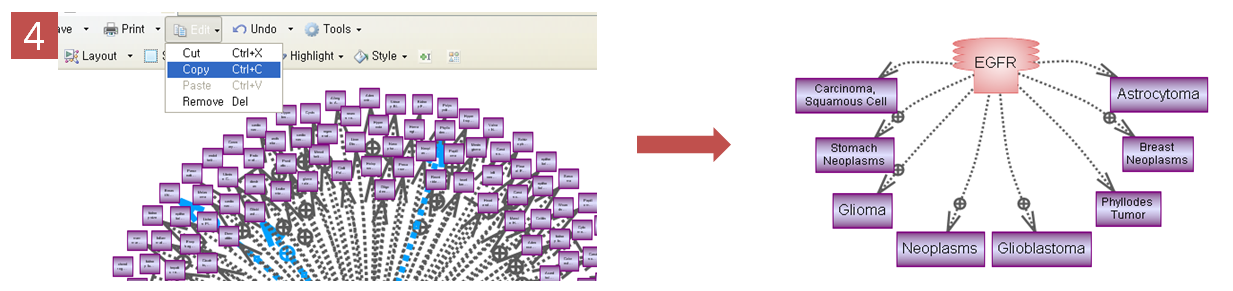
문화가 아름다운 회사 Insilicogen의 CultureDay!
지난 7월에 이어 그 두 번째 시간이 돌아왔습니다.기다리고 기다리던 CultureDay.
이번에 소개할 영화는 줄리아 로버츠 주연의 <먹고 기도하고 사랑하라>입니다.
용기가 필요한 당신을 위한 기적같은 여행!
안정적인 직장, 번듯한 남편, 맨해튼의 아파트까지 모든 것이 완벽해 보이지만 언젠가부터 이게 정말 자신이 원했던 삶인지 의문이 생긴 서른 한 살의 저널리스트 리즈. 결국 진짜 자신을 되찾고 싶어진 그녀는 용기를 내어 정해진 인생에서 과감하게 벗어나 보기로 결심하는데요. 일, 가족, 사랑 모든 것을 뒤로 한 채 무작정 일년 간의 긴 여행을 떠난 리즈는 이탈리아에서 신나게 먹고 인도에서 뜨겁게 기도하고 발리에서 자유롭게 사랑하는 동안 진정한 행복을 느끼고 있는 자신을 발견하게
됩니다. 이제 인생도 사랑도 다시 시작할 수 있을까요?
리즈는 누가봐도 완벽한 조건의 결혼생활을 하고 있는 듯 합니다. 하지만 그녀는 이혼을 결심하게 되고 그 이유는 '자신을 찾기 위해서'였습니다. 힘든 이혼수속을 마치고 그녀는 이탈리아, 인도, 발리로 1년 간의 여행을 떠납니다. 각 여행지마다 만나는 새로운 친구들과 소중한 깨달음 그리고 진정 사랑하는 사람을 만나기까지의 야야기가 영화 속에 담겨 있습니다.
첫 번째 여행지인 로마에서 리즈는 ‘일하는 것 못지않게 중요한 게 놀고 쉬는 것’임을 깨닫습니다. 그녀가 피자 한 입을 베어물며 행복해 하는 장면은 그런 점에서 먹는 즐거움의 진수를 보여주는 것이라 할만하답니다. 그녀가 지금까지 살기 위해 먹어왔다면 오로지 먹는 즐거움만으로도 삶의 이유가 될 수 있다는 것을 보여줍니다.
리즈가 두 번째 여행지로 찾은 곳은 힌두교 성지인 인도의 아쉬람. 어쩌면 로마가 현실적이고 육체적인 욕구를 채우기에 적합한 공간이었다면 아쉬람은 정신적 허기를 채우는데 안성맞춤인 공간입니다. 아쉬람이 제공하는 고요와 응시, 고통과 인내의 메시지들은 충분히 공감을 일으킬만 합니다. 리즈 역시 스스로 선택한 여행지이므로 고되고 고통스러운 시간을 감내할 마음의 준비를 하고 있었겠지만 실제 아쉬람 생활에 힘겨워합니다. 비로소 왜 그녀가 여행을 떠나야 했는지, 그녀의 아픔이 어떤 것이었는지, 그녀가 뉴욕의 일상에서 매우 아파했다는 사실을 알 수 있게 합니다. 이 곳에서 그녀는 자신을 억눌러왔던 상처를 치유하게 되는데요. 단순히 상처를 치유한 것에 그치지 않고 그녀가 명상을 통해 만난 것은 즐거움 이상의 달콤함입니다.
그녀는 그들을 통해서 생각하고 깨닫고 심지어 사랑하는 방법까지 배우게 됩니다. 마지막 종착역인 발리에서는 자신을 돌보는 것을 넘어 남을 돕고 생각한다는 것을 손수 보여줍니다. 그녀의 요청에 고민도 없이 손을 내밀어 주는 그녀의 친구들. 하지만 정작 리즈는 자신의 닫힌 마음을 열 수가 없었습니다. 이제 간신히 명상과 친구들과 규칙적인 생활을 통해 평정을 되찾은 그녀에게 '펠리페'라는 매력적인 남자가 다가오지만 그녀는 사랑에 빠질 수가 없었습니다. 그동안 그녀가 겪었던 슬픔과 시련의 상처가 얼마나 깊었는지를 보여주는 대목이지요.
육체적인 휴식, 정신적인 치유 이외에 그녀에게 필요했던 것은 무엇일까요? 발리를 끝으로 그녀는 여행을 끝냅니다. 그녀가 유지해내려던 것은 삶의 균형입니다. 무엇과 무엇의 균형일까요? 육체적인 것과 정신적인 것의 균형? 너무 사랑에 빠진 삶도 아니고 너무 정신적 사랑만을 강조하는 삶도 아닌 그런 삶의 균형? 영화는 '이것이다'라고 분명히 하지는 않습니다.
하지만 리즈의 고민만큼은 분명하지요. 다시 사랑하는 사람을 만났지만 과연 사랑해도 될 것인지, 그 사랑으로 인해 다시 상처를 입고 아파하지는 않을지, 돌이키고 싶지 않은 자신의 트라우마 때문에 망설이는 그녀의 모습에서 왜 이 여인의 여행이 인도에서 끝나지 않았는지 이해할 수 있게 됩니다. 이 영화는 발리를 끝으로 막을 내리지만, 리즈는 그녀의 새 사랑에게 '함께 건너자'고 합니다. 그리고는 배를 타고 새로운 여행을 떠납니다.
혼자는 균형을 이루기 어려운 법. 어쩌면 두 사람이라야 균형은 완성되는 것일지 모릅니다. 배를 타는 것도 균형을 이루지 못하면 뒤집히고 마는 것처럼, 삶이란 그런 것. 사랑에 완성이란 없는 법. 사랑이란 끊임없이 치우치지 않도록 균형을 맞추는 데 있다는 사실. 삶이란 사랑만도 아니고 그렇다고 사랑만이 아닌 것도 아닌 그런 균형이 아닐까요? 그녀는 그 위태위태한 여정을 다시 떠나고 있습니다.
깊어가는 가을, 섬세하게 감성을 자극하는 영화와 함께한 CultureDay! 극장을 나서며 저는 당장이라도 이탈리아로, 인도로, 발리로 달려가고 싶었습니다. ^____^
이번 CultureDay를 통해 사람이 좋은 회사, 문화가 아름다운 회사임을 다시 한 번 느끼게 하는 계기가 되었답니다. 발리의 주술사 케투가 건네는 "세상을 머리로 계산하지 말고 가슴으로 느껴"라는 충고를 마음에 새기며...
by KM team web publisher Sunsoo
Posted by 人Co

 이번 MOU 체결식에는 (주)인실리코젠의 최남우 대표이사님, 숭실대학교 의생명시스템학부의 학부장님이신 김상수 교수님이 참석하셨으며,
양해각서 체결로 인해 (주)인실리코젠은 5억원 규모의 실습용 생물정보 솔루션과 최신의 생명정보 기술교육 및 현장실습을 지원하기로
하였으며, 숭실대는 최첨단의 하드웨어 시스템과 다양한 인적네트워크를 제공하기로 동의하였습니다.
이번 MOU 체결식에는 (주)인실리코젠의 최남우 대표이사님, 숭실대학교 의생명시스템학부의 학부장님이신 김상수 교수님이 참석하셨으며,
양해각서 체결로 인해 (주)인실리코젠은 5억원 규모의 실습용 생물정보 솔루션과 최신의 생명정보 기술교육 및 현장실습을 지원하기로
하였으며, 숭실대는 최첨단의 하드웨어 시스템과 다양한 인적네트워크를 제공하기로 동의하였습니다. 
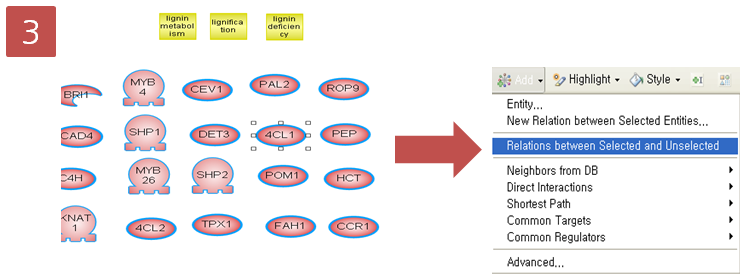
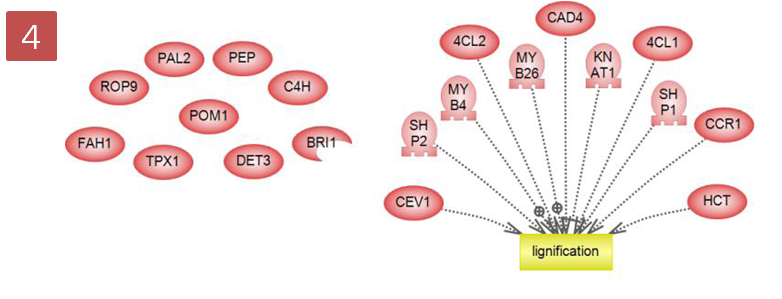
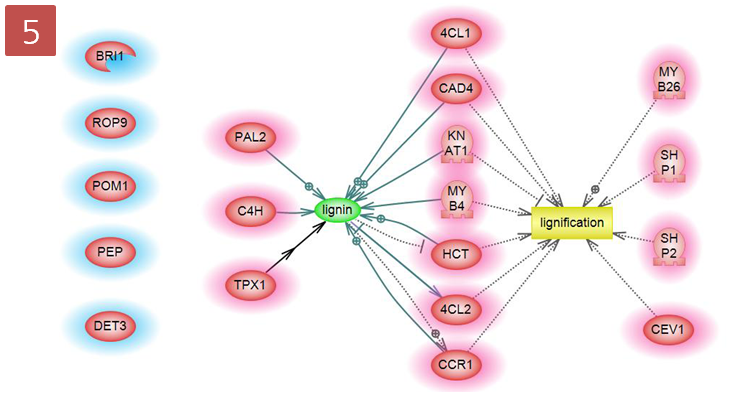
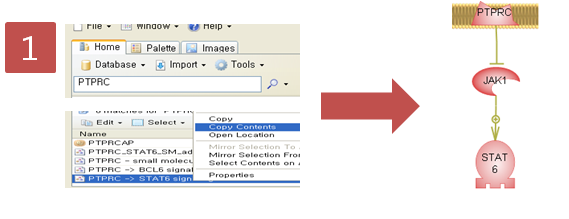
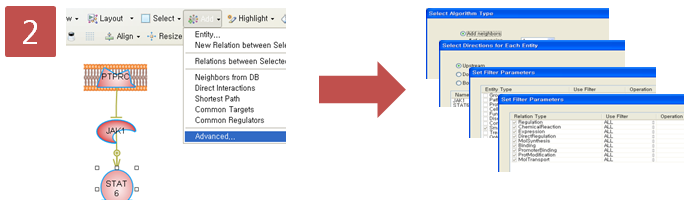
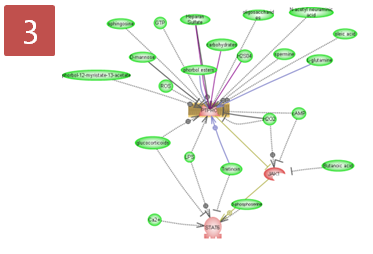
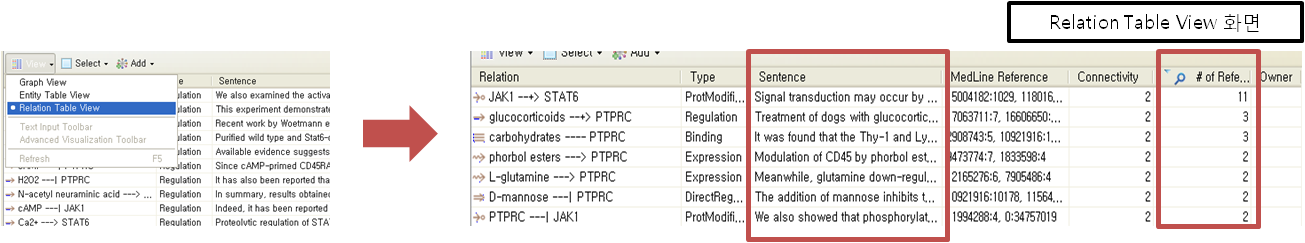
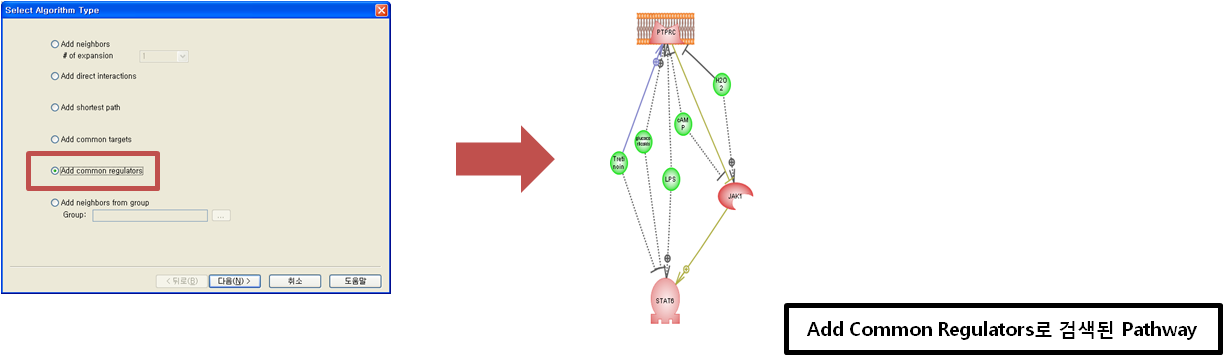

 입사지원서.doc
입사지원서.doc