
A Superior Solution for Microbial Genomics - 3
- Posted at 2016/05/25 17:03
- Filed under 제품소식

우리 몸을 구성하는 세포의 반 이상이 미생물 군집들로 차지하고 있습니다. 따라서 미생물의 분류학적, 유전적 기질은 사람, 동물 그리고 식물의 건강과 밀접한 관계를 가지고 있습니다.
특히 아직은 미생물의 유전적 기능 구성에 대한 정보가 구축되기에 어려움이 있고, 현재 metagenomics 분석 도구들도 기능적 구성이나 샘플간 변화 등을 정확하게 예측하기 위해 노력하고 있습니다. [Lindgreen et al. 2015].
만약 metagenome data를 de novo assemble 할 수 있고, 신뢰할 수 있는 기능 예측 결과를 통해 통계적으로 유의하게 변화된 것을 밝히는 분석도구가 있으면 어떨까요? 이러한 분석 도구가 NGS 데이터의 분석 표준이 되고 미생물 분석을 위해 최적화된다면 연구자분들에게 굉장한 도움을 줄 수 있을 것입니다.
미생물의 metagenomics 분석을 위한 플러그인인 CLC Microbial Genomics Module의 기능과 성능을 확인해 보세요.
결과 정확도
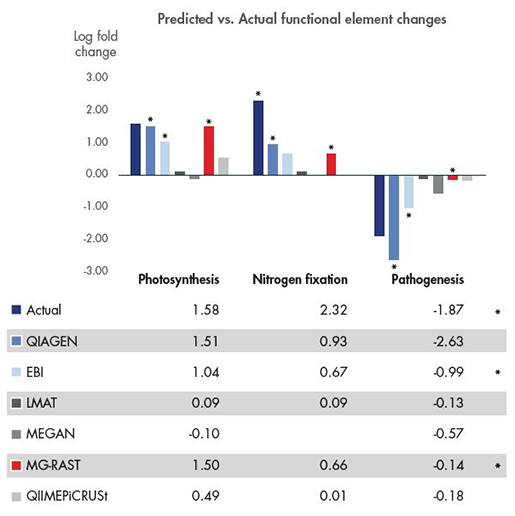
Figure 1. Metagenome 내 높은 정확도의 유전자 기능 예측 및 추적
2016년 1월에 Nature Scientific Reports에 14개의 다른 whole metagenome 분석 도구의 평가 결과에 대해 개재했습니다. 공개된 테스트 데이터를 이용해서metagenome의 기능적 분석이 가능한 5개를 선별하여 CLC Microbial Genomics Module과 비교했습니다. CLC Genomics Workbench에서 제공된 edge 테스트를 이용하여 통계적 분석을 진행하였고, photosynthesis, nitrogen fixation, pathogenesis에 대하여 분석을 진행하였습니다. (*는 통계학적으로 유의한, 정확한 변화를 일관적으로 예측하는 도구를 가리킵니다.)
Metagenomic 데이터를 바탕으로 미생물 군집에서 유전자 기능을 찾는 것은 어렵습니다. 더욱이 다른 metagenome 샘플간의 기능적 성질의 변화를 정확하게 측정하는 것은 더 어렵습니다. QIAGEN 솔루션은 미생물 유전체 분석에서 기능적인 차이를 정확히 찾고 정량화 할 수 있습니다. 또한 샘플간의 통계적으로 유의한 차이를 비교할 수 있도록 해줍니다.
여러 샘플의 비교는 샘플간의 기능적 변화를 찾고, 유사하거나 다른 기능적 요소를 분석하는데 쓰입니다.
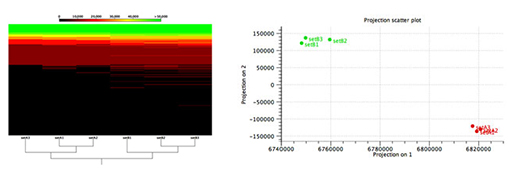
Figure 2: 미생물 샘플들 전반에 걸친 기능적 비교
Metagenome에서 기능적 변화를 찾는 알고리즘은 많이 알려져 있지 않고, 기준이 되는 우수한 모델의 데이터셋이 없기 때문에 어려운 일입니다. 이런 어려움을 극복하기 위해 해당 연구결과에서는 기능을 파악하고 있는 두 합성 미생물 군집으로부터 각각 세개의 데이터셋(A1, A2, A3, B1, B2, B3)들을 만들었습니다.
Figure 2에서 보이는 것과 같이, CLC Microbial Genomics Module은 예측된 기능적 요소들의 비율을 바탕으로 두 개의 군집을 구분 할 수 있습니다.
Metagenome assembly 품질
새로운 Meatgenome assembler에서는 고품질의 어셈블리 결과를 생성하고 유전자 기능을 확인할 수 있습니다.
아래의 Table에서 CLC Microbial Module의 metagenome assembler와 다른 툴에서 misassembly, INDEL, mismatch error 등 다양한 지표들에서 어떤 차이가 나는지 비교해 줍니다.
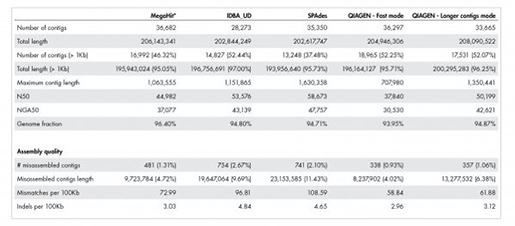
Table 1 : Metagenome assembly의 품질
QIAGEN metagenome assembler는 더욱 정확한 annotation을 가능하게 합니다. 데이터셋의 실제 길이는 209,845,413 base입니다.
실행 시간과 자원 효율성 계산
샘플의 크기가 크거나 데이터의 양이 많을때는 분석 실행시간과 요구되는 리소스가 매우 중요합니다.
테스트 데이터를 가지고 CLC Microbial Genomics Module의 어셈블러와 다른 어셈블러를 비교하였을 경우 분석 시간이 더 짧고 효과적이게 리소스를 이용하는 것을 확인하였습니다.
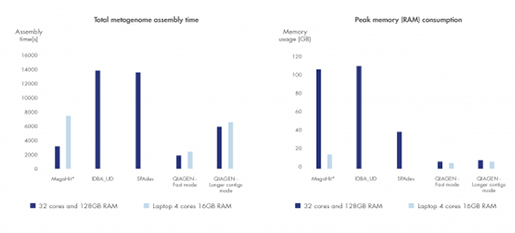 Figure 3. 최고의 metagenome assembly 분석도구
Figure 3. 최고의 metagenome assembly 분석도구
다른 metagenome 어셈블러들과 분석 시간과 리소스 사용면에서 비교하였을 때 우수한 결과를 보였습니다. (*MegaHit는 분석시간을 늘리면서 컴퓨터 메모리 소비를 줄이고 있습니다.)
분석에 소요하는 시간 축소
CLC Genomics Workbench 내의 workflow라는 기능을 이용하면 분석에 소요되는 시간과 노력을 크게 줄일 수 있습니다. 한번에 여러개의 데이터를 넣어줄 수도 있어 분석에 소요되는 시간과 동력을 절감시켜 줍니다.
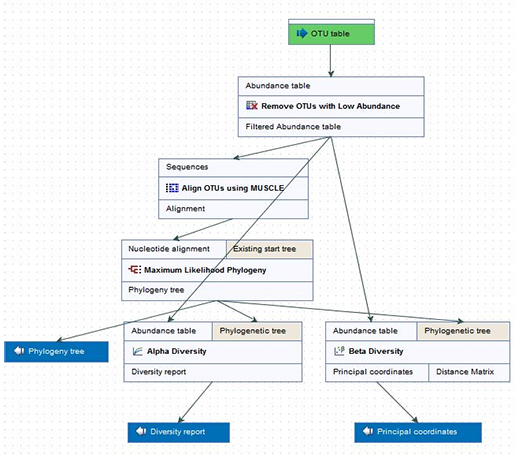
Figure 4. 효율적인 workflow 기능
Posted by 人Co
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/213
Trackback URL : 이 글에는 트랙백을 보낼 수 없습니다

