[Quipu Issue Paper] Bioinformatics Knowledge Management Ⅳ- Gene Network Discovery by Text-mining
- Posted at 2010/04/05 15:44
- Filed under 생물정보
연재 순서
1. Assembly
2. Variation study
3. Expression study
4. Epigenomics
5. Genome Annotation
6. Next Generation Bioinformatics
7. Data Management for web 2.0 Era
8. Semantic Network for Integrated Biology Data
9. Gene Network Discovery by Text-mining
10. Centralization for High-throughput Data Analysis
이번 연재에서는 현재까지 공개 데이터베이스에 축적된 공개된 데이터 및 자신이 보유한 데이터를 이용하여 새로운 정보 및 생물학적 의미를 찾는 Gene Network Discovery by Text-mining에 대해 알아보겠습니다.
3-4. Gene Network Discovery by Text-mining
최근의 생물학 연구의 이슈는 데이터를 생산하는 것보다 현재까지 공개 데이터베이스에 축적된 공개된 데이터 및 자신이 보유한 데이터를 이용하여 새로운 정보 및 생물학적 의미를 찾는 부분에 있다. 즉 데이터의 ‘생산’에서 ‘연결’로 생물정보학의 관점이 옮겨가고 있다고 할 수 있다. 따라서 다양한 분야의 실험데이터, 문헌데이터, 공개데이터 등을 네트워크 형식으로 연결하여 새로운 지식을 발굴할 수 있는 시스템이 주목받고 있다.
현재 NCBI의 PubMed에는 18,000,000건 이상의 논문들이 수록되어 있으며, 하루에도 수 십편의 논문들이 새롭게 업데이트되고 있다. PubMed에 수록되어 있는 저널들은 의학, 생명, 생물에 관련된 연구 논문들이며, 오랜 기간에 걸쳐 저널의 정보에 대한 공신력이 검증된 논문들이라고 할 수 있다. 따라서 연구자가 새로운 주제를 기반으로 하여 연구를 시작하고자 할 경우, 가장 선행되어져야 하는 것은 논문 리뷰라고 할 수 있다. 다른 사람이 비슷한 연구를 수행한 경험이 있는지, 어떠한 방법에 의해서 연구가 진행되었는지, 또한 그 결과는 어떠하였는지 등을 참조하게 된다. 이와 같은 논문 리뷰가 선행된 후 자신의 연구 방향을 설정하게 된다. 그만큼 다른 사람에 의해서 분석되어진 연구 정보가 최근 들어서는 상당히 중요하다고 할 수 있다. 더구나, 최근에는 인터넷을 이용한 데이터의 정보교환이 활발하여 엄청나게 많은 문헌 정보들을 손쉽게 찾아볼 수 있기 때문에 일정한 부분에서는 직접 실험을 하지 않더라도 그와 유사한 실험을 수행한 결과물을 얻을 수 있다. 따라서 이와 같은 문헌 정보의 모래 언덕을 잘 살펴서 보물을 찾아낼 수 있는 방법들이 주목을 받고 있다.
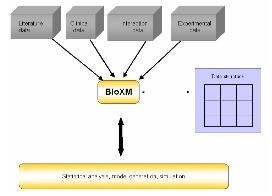
한 예로 Ariadne사의 MedScan과 Pathway Studio는 텍스트 마이닝이라는 컴퓨터 알고리즘을 이용하여 주어진 문헌 정보에서 유전자와 질병, 화학물질, 세포내 프로세스, 대사회로와 같은 엔티티(Entity)들의 관계를 자동으로 추출하여 테이블과 다양한 그래프로 관계들을 보여주는 프로그램으로 대사회로, 유전자 조절 네트워크, 단백질 상호작용 맵과 같은 실험결과를 이해하는데 상당히 유용하다(그림 7). Pathway Studio는 척추동물, 식물 연구의 생물학적 연관관계, ontology와 pathway들의 정보를 포함하고 있는 ResNet 데이터베이스와 자연언어처리기술을 이용하여 과학문헌을 자동으로 읽고 생물학적인 관계를 추출하는 기능을 가진 MedScan으로 구성되어 있다. MedScan의 경우에는 약 1천개의 논문 초록을 대상으로 생물학적인 관계를 추출하는데 2~3분밖에 걸리지 않으므로, 대량의 수집된 논문에서 특정한 바이오마커를 발굴하거나 특정 단백질 또는 질병과 관련된 네트워크 정보를 검토하기에는 상당히 유용하다고 할 수 있다.
보통 하나의 유전자와 관계하는 다양한 정보를 찾아보기 위해서는 수많은 데이터베이스와 문헌, 웹사이트를 검색하여 그 연관관계를 하나씩 도출해야 되지만, Pathway Studio와 같은 프로그램은 그와 같은 일련의 시간과 노동력이 상당히 투자되어야 하는 업무를 효율적으로 지원함으로써 연구자의 보다 빠르고 충실한 결과물을 얻을 수 있도록 지원한다.
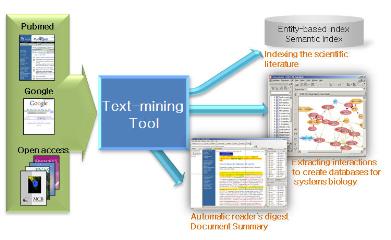
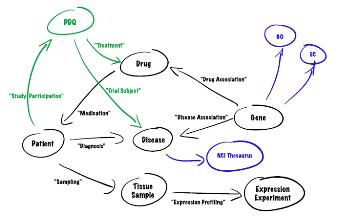
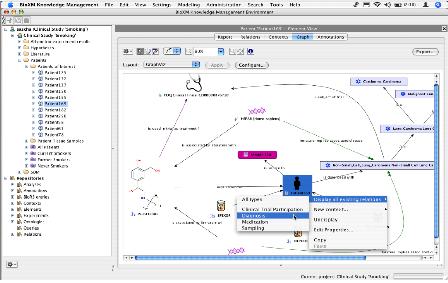
그림 8은 Cholestasis에 관련된 약물과 단백질 등의 연관관계를 Pathway Studio를 이용하여 연구자가 쉽게 이해할 수 있는 방식의 그래프로 재구성한 것이다. 이와 같은 방법으로 복잡한 질병과 약물, 단백질 및 대사 작용의 네트워크를 시각적으로 이해하기 쉽게 제공하고 있다.
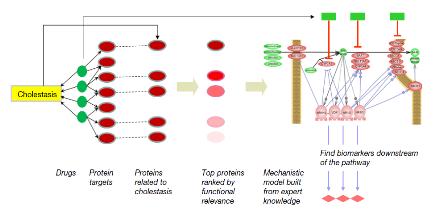 그림 8. Pathway Studio workflow diagram
그림 8. Pathway Studio workflow diagram
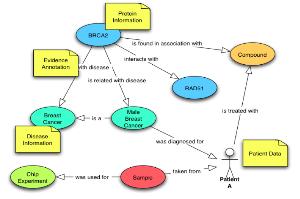
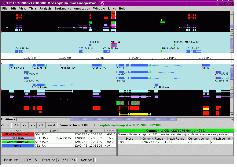
그림 9는 EMB라는 유전자를 대상으로 관련 있는 다양한 유전자 및 질병, 약물, 세포내 프로세스 등을 연결한 그래프로서 연결되어있는 라인을 클릭하면 그림과 같이 연관관계를 표현하는 문헌정보를 확인할 수 있어 연관관계의 정확성 및 신뢰성을 뒷받침하고 있다.
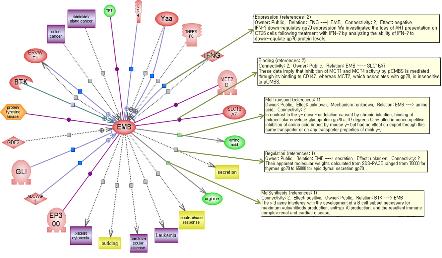 그림 9. Entity와 Relation의 네트워크 및 관련 문헌의 확인
그림 9. Entity와 Relation의 네트워크 및 관련 문헌의 확인
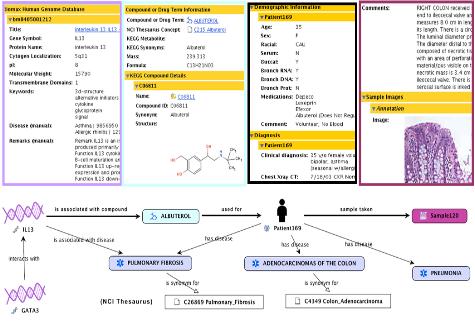
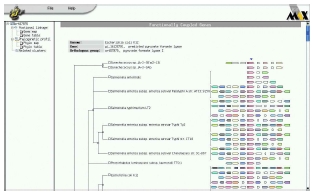
그림 10은 PubMed에서 Curcumin과 Prostate Cancer에 관련된 논문을 검색하여 수집된 수 십 여 편의 논문에서 MedScan의 텍스트 마이닝 알고리즘을 이용하여 네트워크를 재구성한 것이다. 그림에서 보는 것과 같이 Curcumin과 Prostate Cancer 사이에 있는 단백질이 Prostate Cancer를 억제하는 역할을 한다는 정보를 검증된 문헌을 통해서 확인하는 것이다.
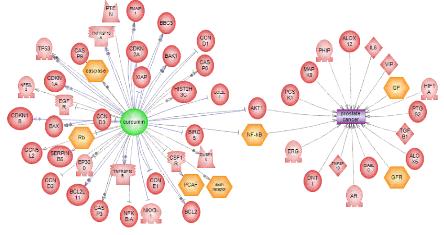 그림 10. MedScan을 통한 문헌정보의 네트워크 구성
그림 10. MedScan을 통한 문헌정보의 네트워크 구성
그림 9는 EMB라는 유전자를 대상으로 관련 있는 다양한 유전자 및 질병, 약물, 세포내 프로세스 등을 연결한 그래프로서 연결되어있는 라인을 클릭하면 그림과 같이 연관관계를 표현하는 문헌정보를 확인할 수 있어 연관관계의 정확성 및 신뢰성을 뒷받침하고 있다.
다음 연재에서는 NGS Edition의 마지막 연재로 대용량의 데이터를 다루기 위한 Centralization for High-throughput Data Analysis에 대해 알아보도록 하겠습니다.
많은 관심 부탁드립니다.
많은 관심 부탁드립니다.
Posted by 人Co
- Tag
- Ariadne, EMB, Entity, insilicogen, MedScan, NCBI, Network, NGS, Ontology, Pathway Studio, PubMed, Relation, ResNet, Text-mining, 문헌, 바이오마커, 엔티티, 인실리코젠, 자연언어처리기술, 텍스트 마이닝
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/67