숭실대학교, (주) 인실리코젠 인재양성 MOU 체결
- Posted at 2011/01/04 19:50
- Filed under 회사소식
지난 1월 3일, (주)인실리코젠과 숭실대학교 의생명시스템학부는 맞춤형 생명정보 인재 양성을 위한 MOU를 체결하였습니다.
 이번 MOU 체결식에는 (주)인실리코젠의 최남우 대표이사님, 숭실대학교 의생명시스템학부의 학부장님이신 김상수 교수님이 참석하셨으며,
양해각서 체결로 인해 (주)인실리코젠은 5억원 규모의 실습용 생물정보 솔루션과 최신의 생명정보 기술교육 및 현장실습을 지원하기로
하였으며, 숭실대는 최첨단의 하드웨어 시스템과 다양한 인적네트워크를 제공하기로 동의하였습니다.
이번 MOU 체결식에는 (주)인실리코젠의 최남우 대표이사님, 숭실대학교 의생명시스템학부의 학부장님이신 김상수 교수님이 참석하셨으며,
양해각서 체결로 인해 (주)인실리코젠은 5억원 규모의 실습용 생물정보 솔루션과 최신의 생명정보 기술교육 및 현장실습을 지원하기로
하였으며, 숭실대는 최첨단의 하드웨어 시스템과 다양한 인적네트워크를 제공하기로 동의하였습니다.
(왼쪽부터 (주)인실리코젠의 최남우 대표이사님, 숭실대학교 의생명시스템학부 김상수 교수님)
이번 협정은 국내 기업과 연구기관에서 요구하는 생명정보 실무 교육을 통해 바이오 연구개발에 필요한 현장중심의 인재를 양성하는 것이
목적이며 이러한 산학협동 과정은 국내 최초로 시도되는 사업이라고 생각됩니다. 또한 생명정보 교육을 한 단계 성숙할 수 있는 계기를
마련하였으며, 빠르게 변화하고 있는 생명정보의 지식과 졸업 후 산업현장에서
활용할 수 있는 전문 능력을 갖출 수 있게 되었습니다.
활용할 수 있는 전문 능력을 갖출 수 있게 되었습니다.
Posted by 人Co
- Tag
- Bioinformatics, MOU, 산학협동, 생명정보, 생물정보, 숭실대, 의생명시스템학부, 인실리코젠
- Response
- No Trackback , 1 Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/89
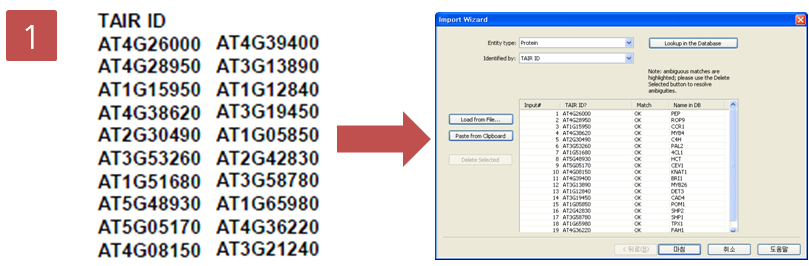
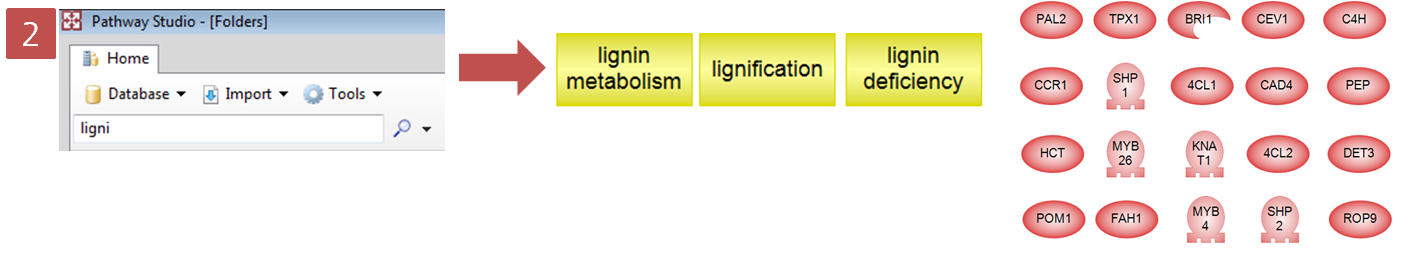
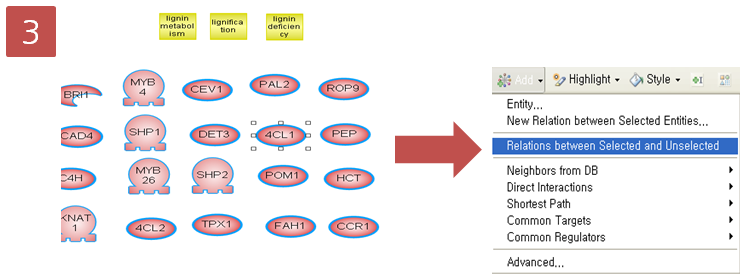
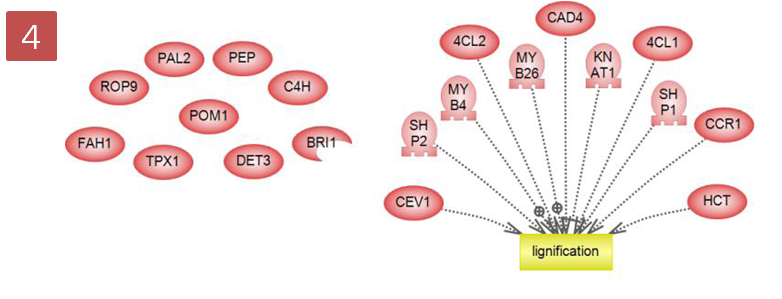
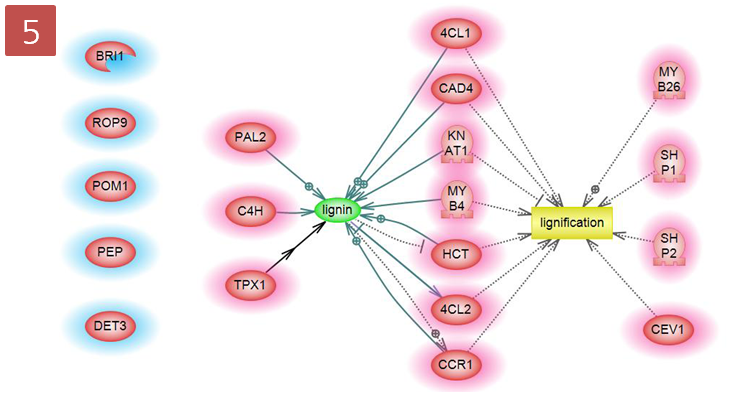
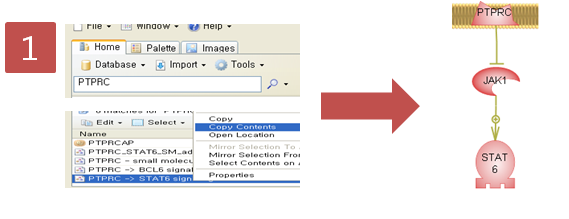
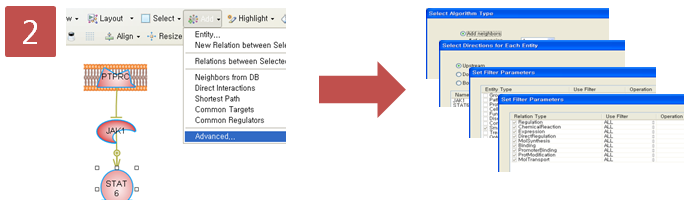
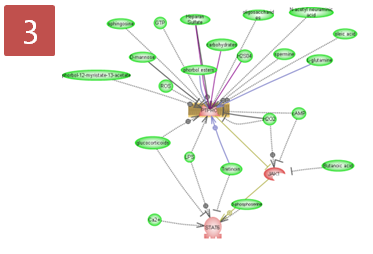

 입사지원서.doc
입사지원서.doc