ChIP-Seq
ChIP 은
Chromatin Immunoprecipitation의 약자로 세포내에서 이뤄지는 단백질과 DNA간의 상호작용을 알아내는 주요한
방법으로 특정 단백질과 binding 하는 DNA sequence 를 알아내는 것을 목적으로 합니다. 특정 단백질과 결합된
DNA을 면역학적 방법인 antibody를 이용하여 침강시킨후 결합된 DNA를 따로 분리하여 그 sequence를 확인합니다.
이때, 해당 서열을 확인 하는 방법으로 microarray방식을 이용하면, ChIP-chip이 되고, NGS와같은 시퀀싱 방식을
이용하면 ChIP-seq이 됩니다. 이러한 방법은 유전자 발현을 조절하는 전사조절인자(transcription factor)의
bindig site와 기작을연구하는데 많이 이용되고 있습니다.
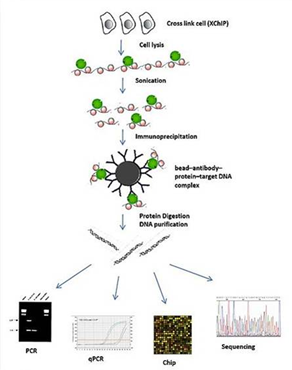
예를 들어 transcription factor A의 binding-site를 분석하기 위해, 먼저 세포내에서
transcription factor A와 DNA를 결합시킨 후 그 결합을 고정하기 위해 sample을 포름알데히드와 같은
고정액으로 고정시킵니다. 이후 세포를 lysis하여 DNA 전체를 분리한 다음 sonication 방법으로 DNA를 잘게
조각냅니다. 그러면 transcription factor와 결합된 상태의 DNA 조각과 그렇지 않은 조각이 생성됩니다. 이후
원하던 transcription factor A에 binding된 DNA 조각만을 분리하기 위해 transcrition
factor A 특이적인 antibody와 beads 붙여 원심분리를 통해 transcription factor A와 이에 결합된
DNA만을 분리해 낼 수 있습니다. 마지막으로 분리된 transcription factorA와 DNA 사이의 결합을 끊어
DNA만을 분리해낸 다음 앞서 언급한 microarray방식과 NGS 기술을 이용한 시퀀싱 방식을 통해 각각 확인할수 있습니다.
Microarray방식은 유전체상의 대부분의 영역을 microarray probe로 제작하여 chip에 심은 후
transcription factor A와 결합되었던 DNA조각을 binding 시켜 확인하게 되며, 시퀀싱 방식은 분리된
DNA조각을 직접적으로 시퀀싱을 통해 확인하게 됩니다. 이후 시퀀싱된 서열을 해당 유전체 서열에 mapping(reference
assembly)을 통해 유전체 상의 binding location을 확인 합니다. 이들 모두 공통적으로 transcription
factor A가 binding 하는 서열정보를 비롯하여 유전체내의 binding location을 함께 확인 할 수 있어
이차적으로 전자를 조절 받는 유전자 프로파일을 함께 확인 할수 있는 이점이 있습니다.
단백질과 binding 되는 DNA 서열이 짧기 때문에 , ChIP-Seq 분석을 할 때는 일반적으로 short read로
시퀀싱을 진행합니다. 또한 reference 서열에 mapping 할 때 역시 mapping 파라메터들을 엄격하게 설정하여
noise data의 생성을 예방합니다. 시퀀싱 reads의 서열들이 짧기 때문에 적은 bp의 mismatch나 gap일 지라도
실제 binding site가 아닌 엉뚱한 위치에 mapping될 확률이 높아 지므로 최종적으로 ChIP peak를 찾기
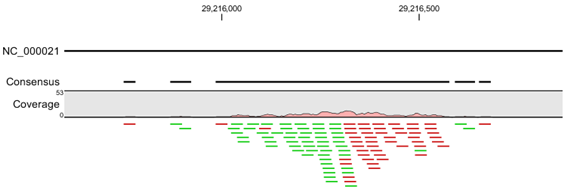
힘들어지게 될 수 있습니다. Mapping view를 보면 이렇게 특정 단백질에 특이적인 binding-site에만 read들이
mapping 되어 형성되는 'peak'을 확인 하실수 있습니다(Candidated transciption factor A
binding position).
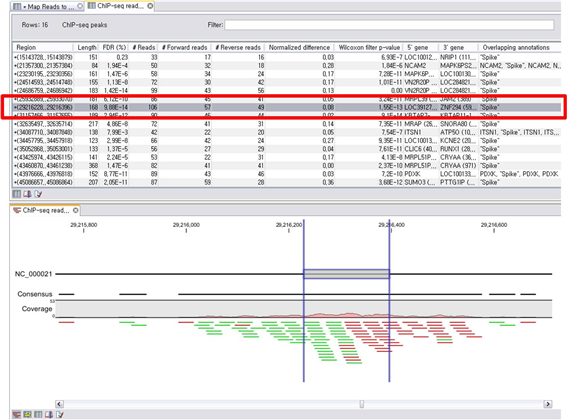
Peak 영역에 mapping된 read의 수와 전체 reference 서열의 mapping 된 read의 분포, 그리고
control 데이터에 mapping된 read의 분포 등을 고려하여 관찰되는 peak가 false positive인지 false
negative인지 통계적으로 유의성을 검증할 수 있습니다. CLC Genomics Workbench를 이용하면 이렇게 찾아진
ChIP peak들에 대한 정보가 담긴 테이블과 해당 ChIP peak가 위치한 부분의 mapping view를 한 화면에서
확인할 수 있습니다.
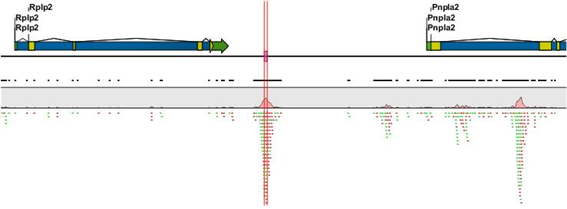
그리고 mapping view를 조금 더 축소해 보면 해당 peak의 upstream과 downstream에 위치한 유전자를
확인하여, 어떤 유전자들이 해당 transcription factor A에 영향을 받을지 유추해 볼 수 있습니다.
부가적으로 BIOBASE사의 'TRANSFAC' 데이터베이스는 발표된 모든 논문들을 대상으로 생물 전문 큐레이터들이 검토하여
transcription factor와 transcription factor binding site에 대한 정보를 축적하고
있습니다. 또한 이렇게 형성된 TRANSFAC의 데이터는 보다 효율적으로 연구자들에게 공급하기위해 CLC Genomics
Workbench에서 plug-in을 통해 ChIP-seq을 통해 찾아진 peak와 직접적으로 비교하여 관련된 유전자, 질병 및
mutation에 대한 다양한 정보를 제공 하고 있습니다.
Posted by 人Co