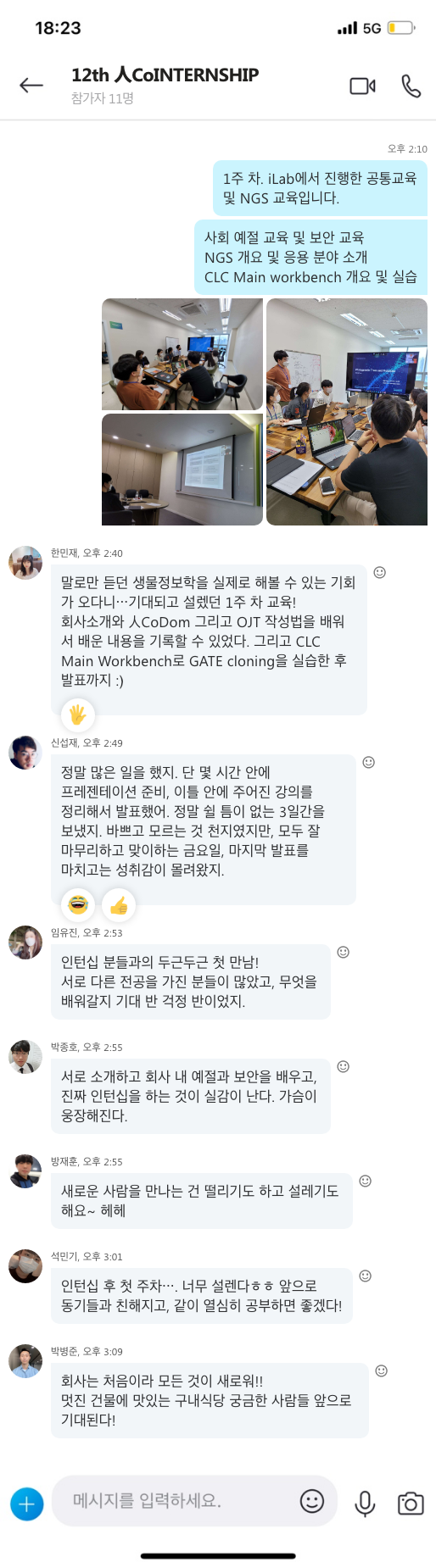
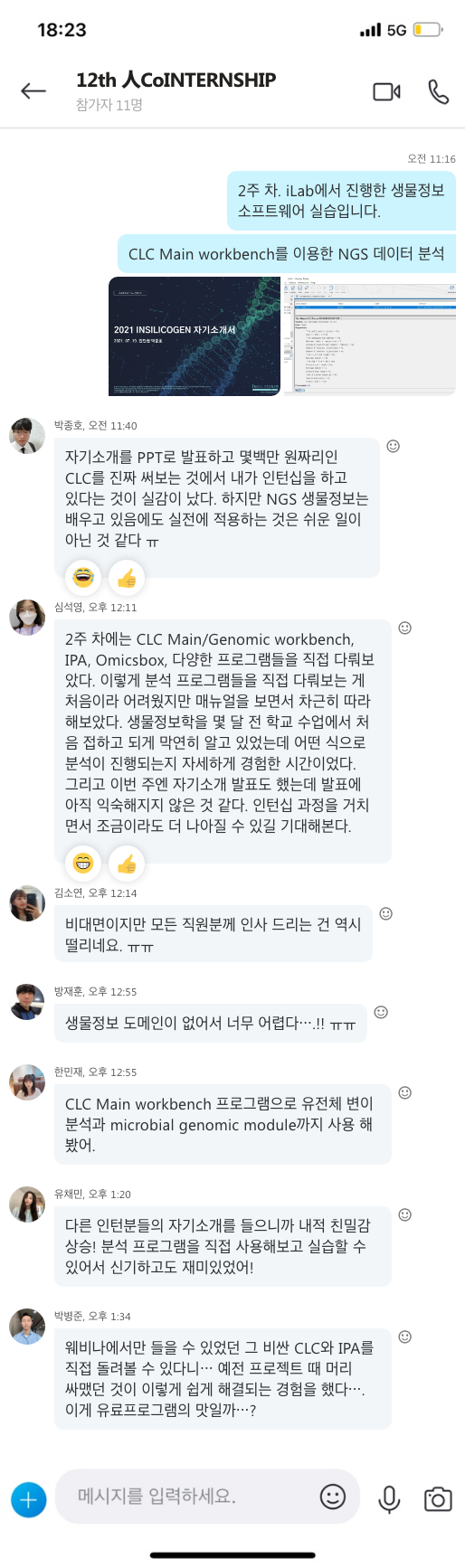
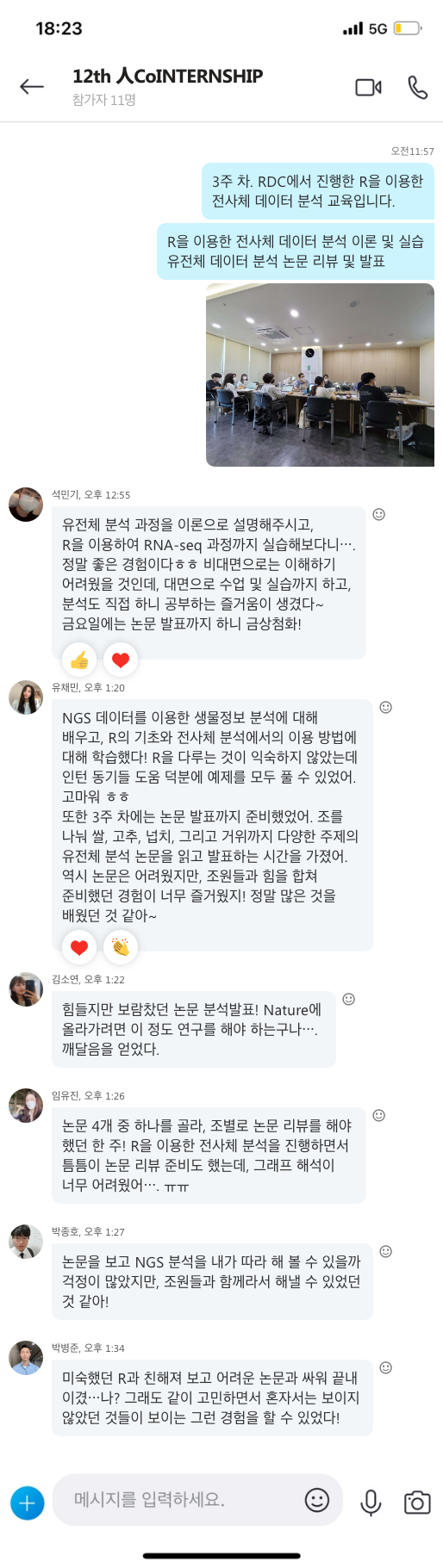
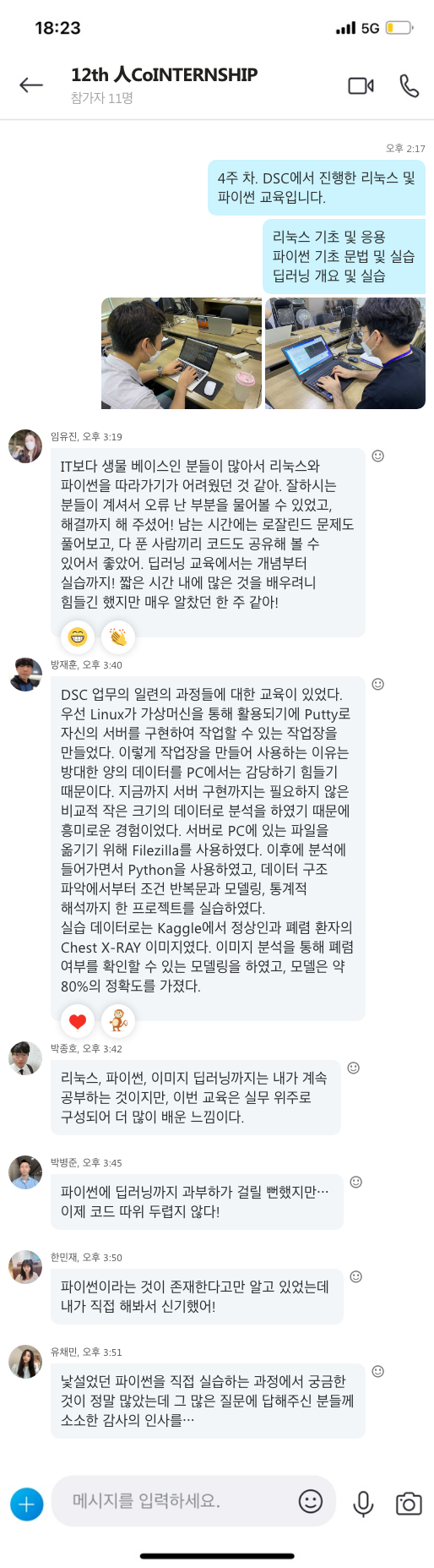
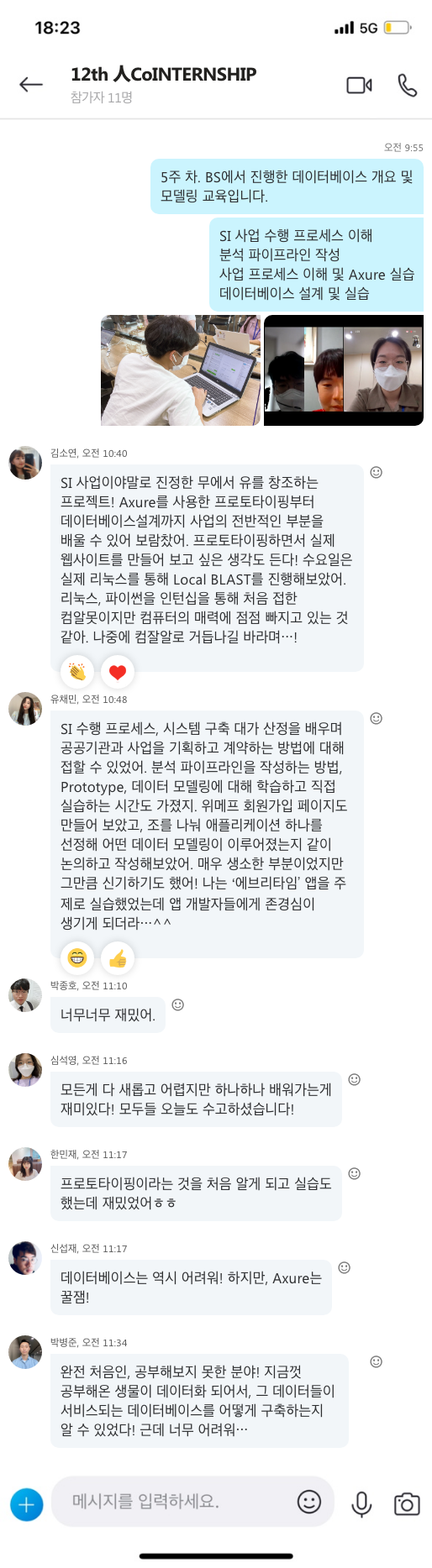
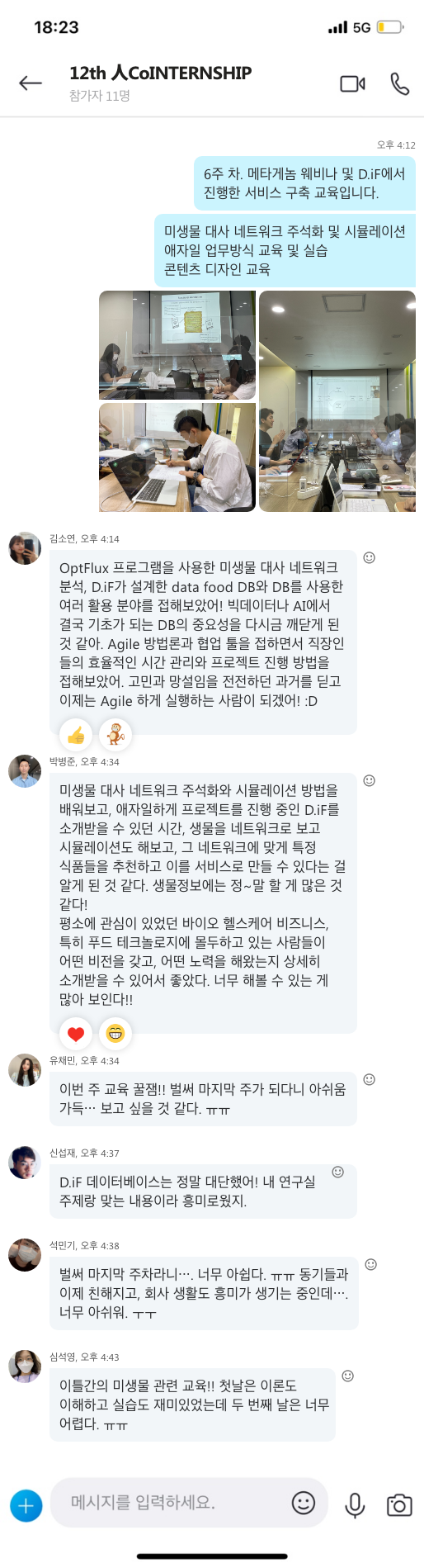
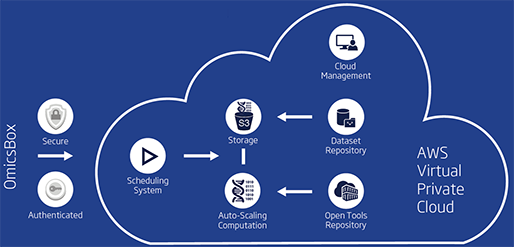
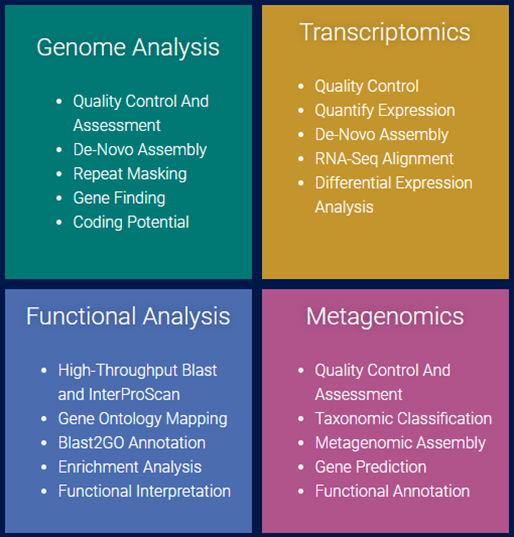
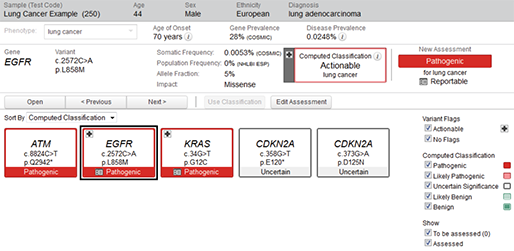
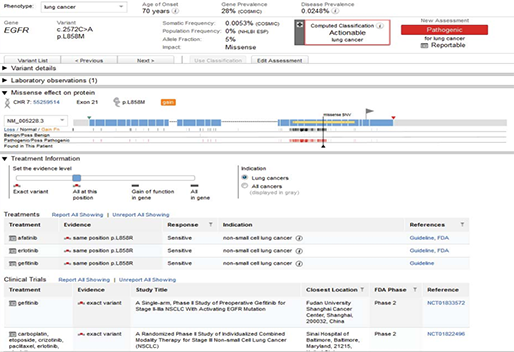
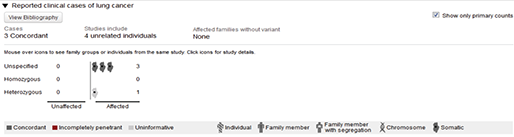
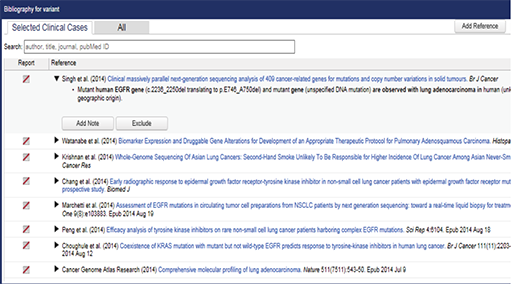
지난 10년간 연구자들에게 많은 사랑을 받으며, NGS 분석을 위한 기초 툴로써 자리를 잡은 CLC Genomics Workbench가 2018년 11월 28일에 12버전으로 정식 릴리즈 되었습니다. 사용자 편의성을 도모하기 위해 많은 부분이 변경되었는데요, 주요한 변화들에 대해서 함께 알아보도록 하겠습니다.
CLC Genomics Workbench 11버전 interface로 변경된 지 4년 정도가 지났습니다. 그리고 이번 메이저 업그레이드에서 CLC Genomics Workbench가 새로운 옷을 입었습니다.
메인화면
전체적인 구성이나 아이콘에는 큰 변화가 없지만, 프로그램을 실행하고 나면 뷰어 화면에 시작하는 방법, 데이터 import를 도와주는 화면이 있으며 예제 데이터도 간단히 다운로드하여 사용하실 수 있습니다. 또한, 자주 사용할법한 도움말들을 뷰어 화면에 배치하여 처음 CLC Genomics Workbench를 사용하는 입문자들도 기존에 비해 접근이 용이하도록 구성했습니다.
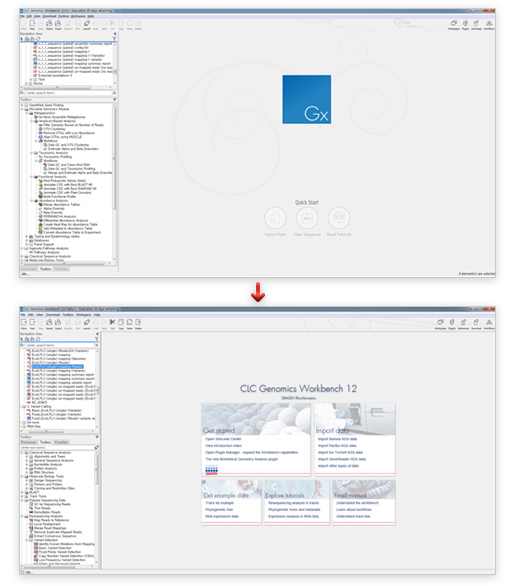
[그림 1] 메인화면의 변화(위-11버전, 아래-12버전)
Import 메뉴의 변경
Import 화면을 보시면 기존의 11버전보다 두 가지 항목이 추가되었습니다. QIAGEN에서 나온 NGS sequencing platform인 GeneReader를 읽을 수 있게 되어 있으며 기존의 Biomedical Genomics Workbench에만 있던 'Import Primer Pairs'가 추가되어 QIAGEN gene panel primer 파일을 바로 가져올 수 있습니다.
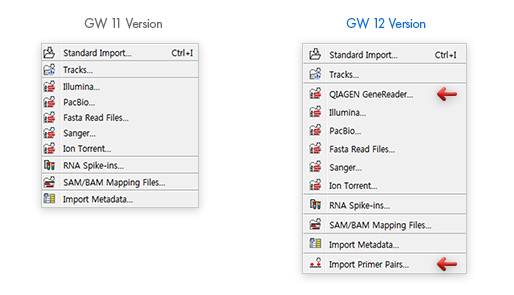
[그림 2] Import 메뉴화면
Navigation Area의 변화
기존 Navigation Area 내에서 파일 혹은 폴더의 순서를 변경할 때, 파일이 생성되거나 옮겨진 순서대로 정렬되어 원하는 대로 정렬하기가 쉽지 않았습니다. 이번 업그레이드 통해 파일이나 폴더를 쉽게 드래그 앤드 드롭으로 순서를 변경할 수 있게 되었습니다. 또한, 상단의 Navigation Area에서 데이터에 마우스를 가져다 대면 뜨던 정보안내 말풍선 창의 정보가 추가되었습니다. 기존 버전에서는 이름만 표시됐던 반면에 12버전에서는 용량과 파일의 포맷을 함께 나타내줍니다.
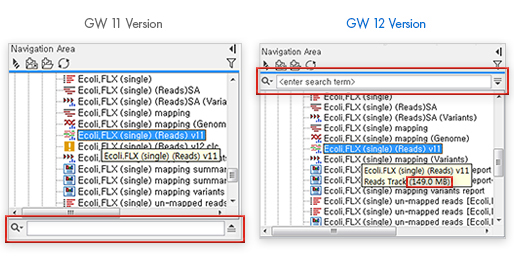
[그림 3] 데이터 타입과 용량 정보 보여주기
자동파일압축
이번 업그레이드에서 놀라운 기능은 기본적으로 압축 기능이 추가되었다는 점입니다. 같은 파일을 동일한 조건으로 분석했을 때, 11버전에서는 149MB였던 결과 파일이 12버전에서는 92MB로 30% 정도 용량을 아낄 수 있게 되었습니다. 기존의 100TB 용량을 이제 130TB처럼 사용하실 수 있습니다.
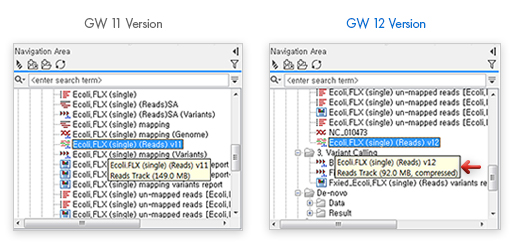
[그림 4] 자동 파일 압축 및 약 30% 저장용량 절약
레퍼런스 데이터 다운로드 방법 변경
기존에는 우측 상단의 Download 아이콘에서 Reference를 선택하여 열리는 창에서 'Download Reference Genome Data'를 다운로드할 수 있었습니다. 새롭게 변경된 UI에서는 Biomedical Genomics Workbench처럼 우측 상단의 'References'라는 아이콘을 이용하여 레퍼런스 데이터를 다운로드할 수 있습니다. Reference 아이콘을 누르면 하단의 그림 중 아래 화면 같은 창이 뜨게 되며 여기서 원하는 종 혹은 원하는 데이터를 골라 다운로드합니다.
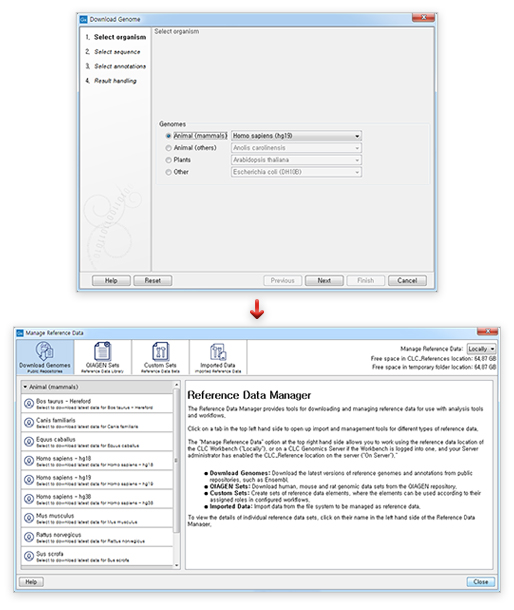
[그림 5] 레퍼런스 데이터 다운로드 인터페이스 변경
Toolbox의 구성 변경 1
plug-in로 제공됐던 'Bisulfite Sequencing'툴이 별다른 설치 없이 기본적으로 탑재 되었습니다.

[그림 6] Bisulfite Sequencing 분석폴더 디폴트로 추가
Toolbox의 구성 변경 2
툴박스의 폴더 구성을 보시면 'NGS Core Tools'가 사라지고, 'Prepare Sequencing Data'라는 폴더에 trimming이나 demultiplex 관련한 툴들이 배치되어 있으며, 'Installed Workflow'로 기존의 'Workflow' 폴더의 이름이 변경되었으며, 'Utility Tools'라는 폴더가 추가되었습니다.
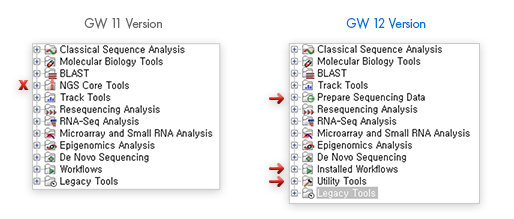
[그림 7] 분석폴더의 재구성
Toolbox의 구성 변경 3
새롭게 추가된 툴에 대해서 소개해 드리면 copy number variant를 분석할 수 있는 툴, variant에서의 정보를 제거하는 부분, RNA-seq을 진행할 때 두 그룹일 때에는 별다른 metadata 없이 진행할 수 있도록 'Different Expression in Two Groups'가 추가되었으며 plug-in으로 사용하던 'Batch Rename'이 추가되었습니다.

[그림 8] 추가된 새로운 툴
몇 개의 툴들은 이름이 변경되었습니다. [그림 9] 이미지를 참고해주십시오.

[그림 9] 이름이 변경된 툴
QIAseq panel reference 다운로드
Reference Data를 다운로드하는 곳으로 가보면 두 번째 아이콘에 QIAGEN Sets라는 아이콘이 있습니다. 이 아이콘을 선택하면, QIAseq Panel에 관련된 reference만 선택적으로 다운로드할 수 있습니다. QIAGEN의 panel을 가지고 나온 데이터를 보다 더 쉽게 분석할 수 있도록 CLC Genomics Workbench에 적용하였습니다.
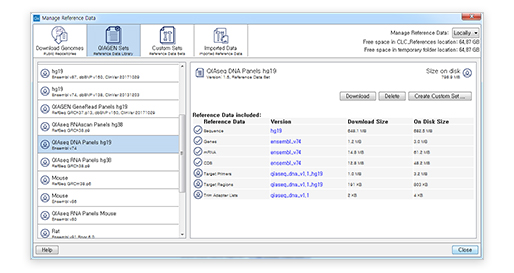 [그림 10] QIAseq 분석에 찰떡인 QIAGEN Sets 다운로드
[그림 10] QIAseq 분석에 찰떡인 QIAGEN Sets 다운로드
손쉬운 서버 프로그램과의 연동
Workbench desktop 버전과 server의 연동에 관련된 부분입니다. 창의 아래쪽 표시줄에 보면 S라고 되어있는 사각형 아이콘이 생성되어 있습니다. 이 버튼을 누르면 Server Connection 창이 뜨게 되고 쉽게 server와 연결할 수 있고 연결 상태를 하단에서 바로 확인할 수 있습니다.
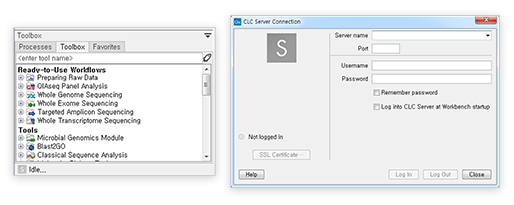
[그림 11] 메인화면에서 연결되는 CLC Server Connection
Track 뷰어의 변화
아래 화면([그림 12])은 read mapping의 track 화면입니다. Track에서 position을 보여주던 숫자가 위치하고 있던 맨 윗부분 위로 크로모좀 뷰어가 추가되었습니다. read 색깔은 unpair/pair 그리고 mismatch까지 색상 지정을 자유롭게 하실 수 있으며 aligned read의 하단에 있던 overflow graph가 read 상단으로 올라와 새로운 coverage graph를 보여줍니다. 그와 동시에 read를 검토할 때 불편했던 위아래 이동에 스크롤바가 추가되어 편하게 read의 alignment를 살펴볼 수 있습니다.
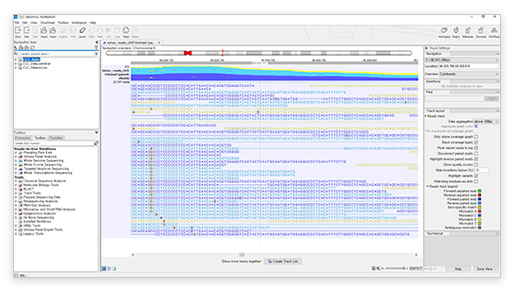
[그림 12] Intuitive 하게 변경된 Track 뷰어 인터페이스
Differential Expression for RNA-seq툴의 개선
기존 버전에서 불가능했던 RNA-seq에서 normalization 방법도 설정할 수 있습니다. 기본적인 whole transcriptome RNA-Seq과 targeted RNA-seq, Small RNA 분석을 따로 설정할 수 있으며 normalization도 TMM과 Housekeeping gene으로 가능합니다.
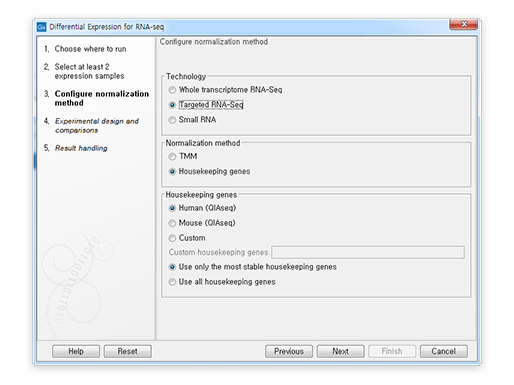
[그림 13] 세분화된 차등발현 유전자 분석 툴
Differential Expression in Two Group 추가
두 그룹 간의 RNA-seq 비교일 경우 별도의 metadata 없이 control과 study 그룹을 wizard에서 설정하여 분석할 수 있도록 구성되어 있습니다.
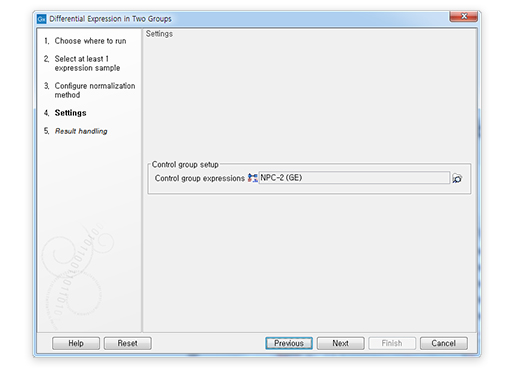
[그림 14] 두 그룹간의 비교 화면
Export 기능의 개선
Annotation을 export할 때 생기던 에러가 해결되었습니다. 기존에 엑셀 포맷으로 export 할 경우에는 모든 칼럼을 전부 export 하던지 혹은 필요한 칼럼만 체크해서 export를 할 수 있었습니다. 현재 내가 보고 있는 화면대로 원하는 칼럼만 export 하려면 다시 체크해야 되는 번거로움이 있었습니다. CLC Genomics Workbench 12버전 업그레이드를 통해 현재 보고 있는 칼럼만 그대로 export 하는 기능이 추가되어 다시 골라야 하는 번거로움을 없앴습니다.
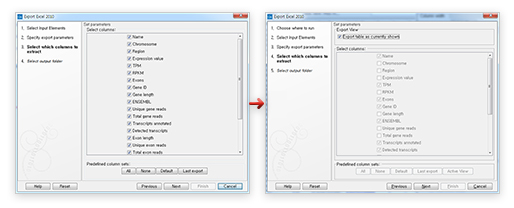
[그림 15] 화면 그대로 간편하게 export 하기
이번 업그레이드로 많은 부분이 바뀌게 되었습니다. 기대하셨던 부분이 반영되었을 수도 있고, 아직 부족한 부분도 있을 거라고 생각이 됩니다. CLC Genomics Workbench는 사용자의 많은 의견을 반영하여 10년 넘도록 꾸준히 업그레이드 되고 있습니다. 업그레이드된 12버전을 통해 새로운 기능들을 확인해보시기 바랍니다.
구버전의 CLC Genomics Workbench를 이용하시는 분 중 12버전으로 업그레이드가 필요하신 분들은 12월 한 달 동안 진행되는 연말 프로모션을 적극 활용하시기 바랍니다.
Posted by 人Co