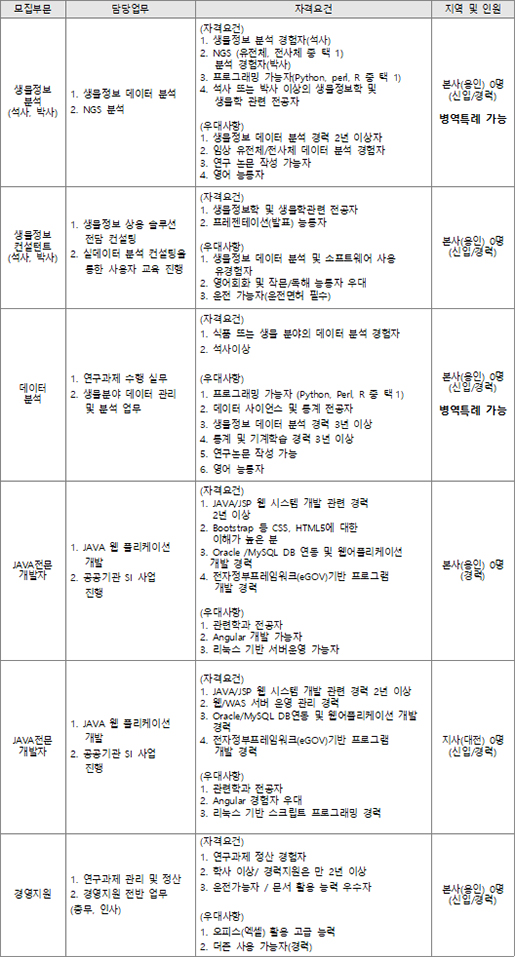
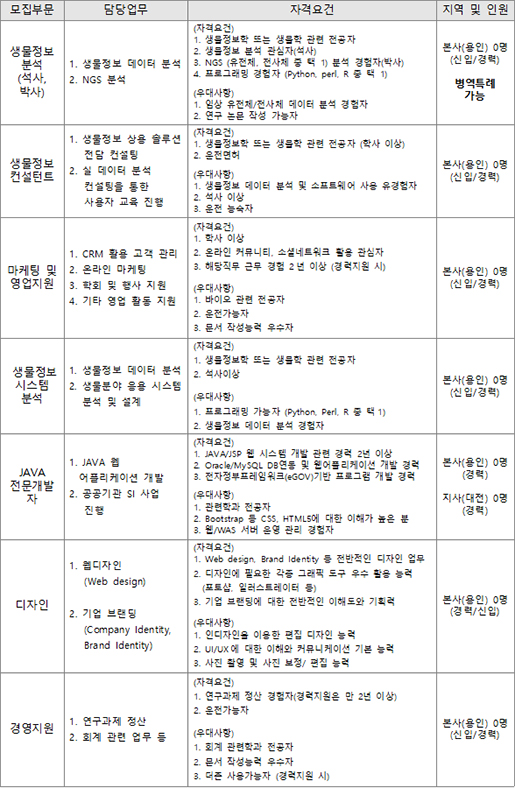
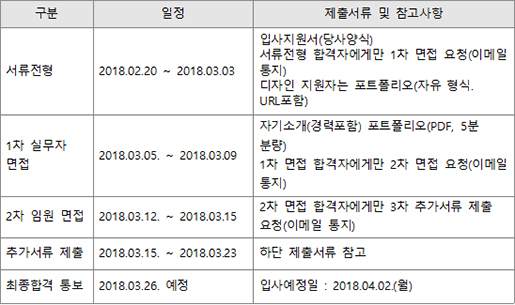
人CoPLAY 2018
- Posted at 2018/05/10 10:51
- Filed under 회사소식
2018년 4월 27일 봄바람 솔솔 부는 따뜻한 봄날 (주)인실리코젠은 '人CoPLAY 2018'을 개최했습니다.

하루를 꼬박 가득 채웠던 작년 단합대회와는 달리,
이번 단합대회는 극심한 미세먼지를 피해 '人CoPLAY 2018 볼링'으로 대체되었습니다.

볼링은 밥심이죠! 오전 근무를 마친 뒤 회사 지하 식당에서 점심 식사 후 볼링장으로 이동했습니다.

팀 구분을 위한 손목 아대가 있는데요, 볼링 시작 전에 손목아대 안에 엑스맨 표식을 표시하여 나누어 드렸습니다.
(두근)(두근) 혹시 내가 엑스맨~?!!

'저는 볼알못(볼링을 알지 못하는 사람들)과 엑알못(엑스맨을 알지 못하는 사람들)인데요!'
하시는 분들을 위한 '人CoPLAY 2018 포토제닉’ 도 실시하였습니다.
'人CoPLAY 2018’ 사진을 응모받아, 심사 후 1,2,3등 분들께 상품권을 지급해드렸습니다.

볼링경기를 마친 뒤 함께 맛있는 소고기를 먹으러 갔습니다.
사장님의 개회사와 함께 '人Co PLAY 2018 볼링’, ‘엑스맨을 찾아라’ 시상식을 진행하였습니다.

대망의 1위 팀은?!!
(두구)(두구)(두구)(두구)
축하드립니다. 노랑팀!
팀전에서는 노랑팀이 최종 우승을 하였네요.
참여하신 모든 분들 모두 모두 축하드립니다! (짝짝짝)

하루를 꼬박 가득 채웠던 작년 단합대회와는 달리,
이번 단합대회는 극심한 미세먼지를 피해 '人CoPLAY 2018 볼링'으로 대체되었습니다.
볼링은 밥심이죠! 오전 근무를 마친 뒤 회사 지하 식당에서 점심 식사 후 볼링장으로 이동했습니다.
'人CoPLAY 2018 볼링’ 룰은요?
'人CoPLAY 2018 볼링’은 팀전, 개인전으로 나뉩니다.
팀전은 팀 합산 기록이 가장 높은 팀이, 개인전은 남녀 개인이 가장 높은 점수를 기록한 사람이 이기는 룰입니다.
하지만 이렇게 당연한 룰만 있다면 너무 심심하겠죠? (찡긋)

팀 구분을 위한 손목 아대가 있는데요, 볼링 시작 전에 손목아대 안에 엑스맨 표식을 표시하여 나누어 드렸습니다.
(두근)(두근) 혹시 내가 엑스맨~?!!
'저는 볼알못(볼링을 알지 못하는 사람들)과 엑알못(엑스맨을 알지 못하는 사람들)인데요!'
하시는 분들을 위한 '人CoPLAY 2018 포토제닉’ 도 실시하였습니다.
'人CoPLAY 2018’ 사진을 응모받아, 심사 후 1,2,3등 분들께 상품권을 지급해드렸습니다.
볼링경기를 마친 뒤 함께 맛있는 소고기를 먹으러 갔습니다.
사장님의 개회사와 함께 '人Co PLAY 2018 볼링’, ‘엑스맨을 찾아라’ 시상식을 진행하였습니다.
대망의 1위 팀은?!!
(두구)(두구)(두구)(두구)
축하드립니다. 노랑팀!
팀전에서는 노랑팀이 최종 우승을 하였네요.
참여하신 모든 분들 모두 모두 축하드립니다! (짝짝짝)
그간 바쁜 업무로 인해, 못다 한 이야기를 나누며, 단합대회가 마무리되었습니다.
오늘 단합대회는 한마음, 한뜻으로 같은 목표를 향해 나아갔던 뜻깊은 행사였습니다.
행사 준비위원으로서 '人Co PLAY’를 준비했는데요. 참여하시는 모든 분들 인코인답게, 즐겁게 행사에 임해주셔서 부상자 없이 안전하게 마무리할 수 있었던 것 같습니다.
내년에도 즐겁고, 안전하게 즐길 수 있는 '人CoPLAY 2019’를 기대하며 이만 마무리하겠습니다.
행사에 참여하신 인코인 분들 모두 수고하셨습니다.
마치며...

하윤희
1년간의 경험을 통해 올해는 좀 더 효율적인 준비와 진행이 되었던 것 같아요. 아쉬운 점이 남지만, 이 또한 다음 행사를 위한 좋은 경험이었다고 생각합니다.
이번 人CoPLAY는 그 어느 때보다 모두가 함께 즐긴 행사였고, 한걸음 물러나 바라보는 것이 아닌 서로 알려주며 함께 하는 모습이 너무나도 아름다웠습니다.
각자의 역할을 훌륭히 소화한 브랜드위원들과 먼데이트리오 분들께 이 자리를 빌려 덕분에 즐거운 시간이 되었음에 감사드립니다.
또한, 그 시간을 함께 즐겨주신 모든 人Co인들에게 감사의 인사를 드립니다.
용승천
점심식사 : 다 같이 모여서 화기애애한 분위기와 함께 식사한 것이 좋았습니다.
자유시간 : 농구, 카페, 산책 등 다양한 활동을 하여 즐겁게 보내 좋았습니다.
볼링 : 볼링장 이동 시 통제를 하여 정확하게 제시간에 모였으면 더욱 좋았을 텐데 지각 인원이 많아 아쉬웠습니다.
회식 : 회식 장소에서는 다양한 음식을 마음껏 즐길 수 있어 좋았습니다. 또한, 자유롭게 어울려 이야기할 수 있는 분위기가 좋았습니다.
박원
이번 人CoPLAY를 통해 모든 人Co인들이 한자리에 모였다는 것이 큰 의미가 있는 것 같습니다.
오랜만에 서로 인사하고 함께 볼링을 치며 시간을 보내어 즐거웠습니다.
또한, 지난 단합대회에 비해 남,여 모두 쉽게 참여할 수 있는 종목이어서 적극적으로 하려는 모습이 많이 보인 점이 특히 좋았습니다. 이 시간을 준비해주신 브랜드위원회 분들과 먼데이 트리오분들께 다시 한 번 감사드립니다.
경동수
이번 人CoPLAY는 오후 시간을 이용해 자율시간과 볼링시합을 진행하였습니다. 이번 행사는 브랜드위원회와 먼데이트리오 각 인원마다 역할을 부여하여 준비하였습니다. 업무 중 회의시간을 줄일 수 있었습니다. 또 처음으로 임직원 설문조사를 통해 행사 내용과 일정을 잡았습니다. 다수의 의견을 반영할 수 있어 만족감도 증가되었다고 생각합니다. 모두가 책임감 있게 임해주셔서 무사히 행사를 마무리할 수 있었습니다.
진효빈
어느덧 두 번째 준비하는 '人CoPLAY 2018' 이네요. 두 번째 준비하는 人CoPLAY인 만큼 함께 준비하는 브랜드위원회 분들과 합이 맞아서 조금이나마 편하게 준비할 수 있었던 것 같습니다. 개인 업무하랴 인코플레이 준비하랴 바빴던 한 달이 지나가네요. 고생한 만큼 인실리코젠 임직원분들이 즐겁게 하루를 보내신 것 같아 다행입니다. 준비하시는 분들 모두 고생하셨습니다.
작성자 : 브랜드위원회 진효빈
Posted by 人Co
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/280

 입사지원서_성명_지원부문_20180000.docx
입사지원서_성명_지원부문_20180000.docx
 입사지원서_성명_지원부문_20180000.docx
입사지원서_성명_지원부문_20180000.docx