Circos(
http://circos.ca/)는 데이터를 시각화하는 Perl 기반의 소프트웨어 패키지로써 circular layout을 이용한다는 특징이 있습니다. 다양한 track과 plot 타입을 사용할 수 있어 변이나 genomic interval간의 관계를 표현하는데 유용하기 때문에 근래 유전체 분석 및 비교 유전체 분석 논문에서는 필수 피겨로 자리매김하고 있습니다. [그림 1]은 염색체 간 서열의 유사성을 두 가지 map으로 표현하고 있는데 왼쪽은 일반적인 genetic map의 형태로써 모든 염색체를 하나의 그림에 표현하기 어렵고 1:1 비교만 표현할 수 있다는 단점이 있는 반면 오른쪽의 Circos map은 1:1 비교뿐만 아니라 모든 염색체 간 서열의 연관 관계를 표현할 수 있다는 장점이 있습니다.

그림 1. Genetic map과 Circos의 비교
또한 Circos는 [그림 2]에서 보는 것처럼 line, scatter plot, histogram, text, heat map, tile 등 다양한 형태의 plot type을 제공하고 있고 online tutorial (
http://circos.ca/documentation/tutorials/) 페이지에서 각각의 예제 및 사용법을 보여주고 있습니다. 따라서 연구자는 자신이 가지고 있는 데이터의 속성에 따라 가장 잘 맞는 plot type을 지정하여 사용하면 됩니다. 예를 들어 유사한 genomic region 및 유전자의 관계를 표시하고자 할 때는 link type으로, 각 유전자는 tile 또는 text type을 이용하여 표현할 수 있습니다. 또한 expression value, frequency, coverage와 같이 각 위치에 대한 값을 다르게 갖는 경우에는 histogram이나 line plot을 이용하시면 좋습니다.
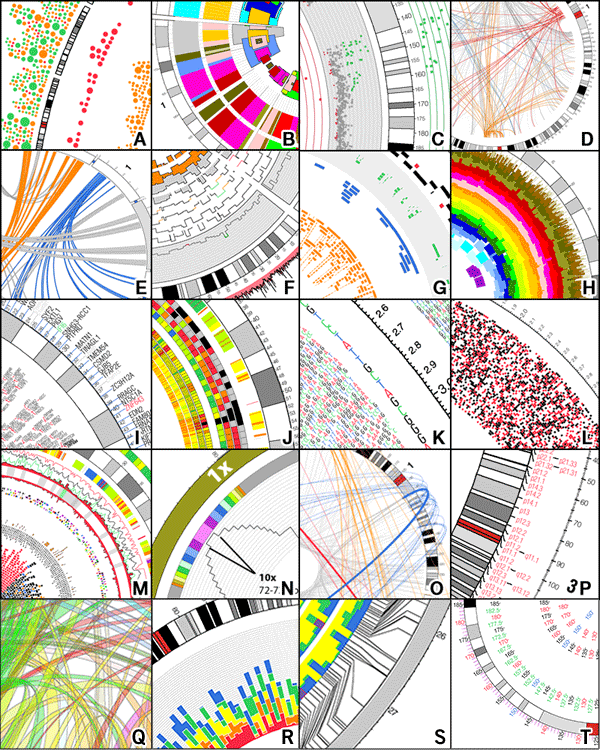
그림 2. Circos의 다양한 plot types
이처럼 그림으로는 너무 친근한 Circos, 말로도 쉬운 Circos, 그러나 막상 그리려니 막막한 Circos, 친해지기는 어려웠던 Circos에 대해서 직접 사용해본 결과 간단하게나마 정보를 드리고자 합니다. Circos map을 그리기 위해서는 [표 1]과 같이 4 단계의 과정을 수행해야 합니다.
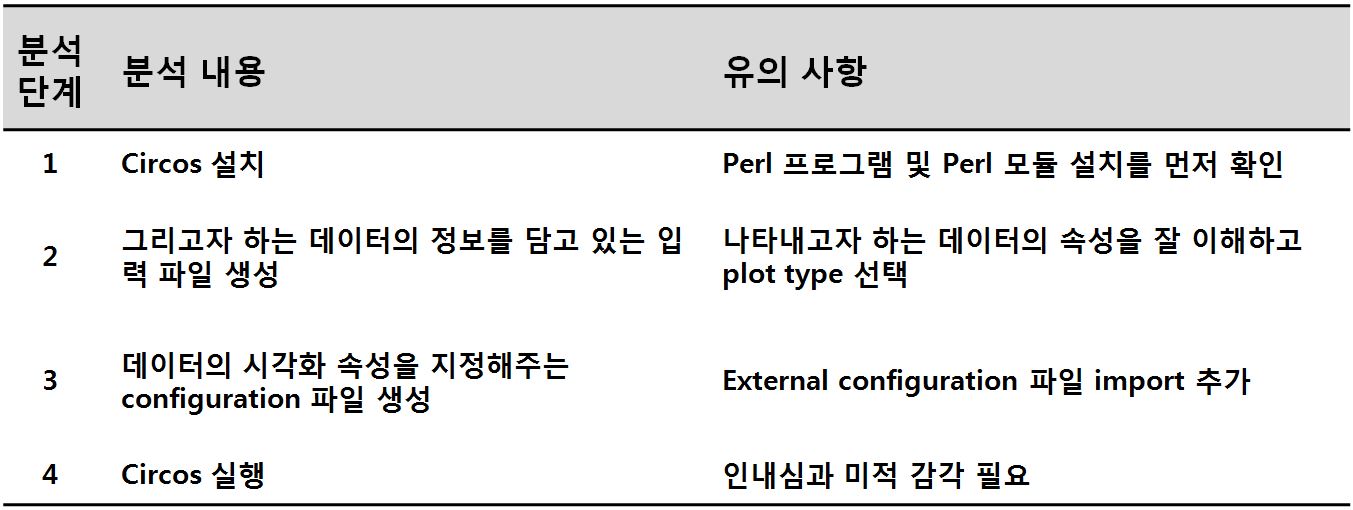
표 1. Circos 분석 워크플로우
1. Circos 설치
Circos의 설치는 비교적 간편한 편으로 홈페이지 (http://circos.ca/documentation/)에서 압축 파일을 다운로드 받은 뒤 압축을 풀어주기만 하면 완료됩니다. 그러나 Circos가 Perl 기반의 소프트웨어이므로 Perl 프로그램 (버전 5.8 이상) 및 분석에 필요한 Perl 모듈 설치가 선행되어야 하고, 그리고자 하는 데이터 각각에 대해 input file 및 별도의 configuration 파일을 작성해 주어야 하므로 처음 접하시는 분들께는 조금 막막할 수 있습니다. 하지만 온라인 tutorial 및 별도의 tutorial 파일을 통해 example data 및 configuration 파일을 함께 제공하고 있으므로 적절히 값을 변경하기만 하면 충분히 응용이 가능하여 조금만 살펴보시면 누구나 쉽게 따라할 수 있을 것입니다.
2. 그리고자 하는 데이터의 정보를 담고 있는 입력 파일 생성
데이터를 어떻게 보여줄 것인지에 따라서 plot type이 결정되고 이것은 Circos map에서 하나의 데이터 트렉을 형성합니다. 데이터 트렉의 위치는 사용자가 직접 지정해줄 수 있으며 여러 데이터 트렉이 중첩되어 더욱 정교한 map을 형성할 수도 있습니다. Circos input file은 이 데이터 트렉에 따라 포맷이 조금씩 달라질 수 있으나 대부분 chromosome (sequence) name, start position, end position, values (color나 값 지정) 순으로 Circos map 상 위치 정보를 가지고 있습니다. 주의할 점은 동일한 plot type을 갖는다 할지라도 데이터 트렉 위치 및 기타 시각화 속성 값이 다르면 데이터 트렉을 따로 생성해야 한다는 점, 즉 input file을 분리 생성해야 한다는 것입니다. 자세한 데이터 설명 및 input file 포맷은 Circos tutorial(http://circos.ca/documentation/tutorials/configuration/data_files/)을 참고하세요.
2-1. Karyotype 설정
Circos를 그리기 위해서 가장 필요한 것은 각 데이터 트렉의 범위를 지정하는 backbone을 설정하는 것입니다. 이는 하나의 염색체일 수도 있고 여러 염색체로 구성된 genome 서열이거나 또는 sequence contig 및 clone일 수도 있습니다. 이러한 backbone에 대한 정보를 담고 있는 것이 Karyotype 파일이며 그리고자하는 genome이나 chromosome의 이름, 크기, 색 등을 지정해 줍니다. 앞으로 그리게 될 모든 데이터는 이 Karyotype 파일이 지정하는 범위 내에서 허용되므로 잘 설정해 주어야 하며 human, mouse, rat, drosophila genome에 대해서는 이미 tutorial에서 제공하고 있으니 쉽게 이용할 수 있습니다. Karyotype 파일에 입력한 모든 서열 정보가 Circos map에 표현되는 것은 아니고 3의 configuration 파일에서 지정해 주어야 표현이 됩니다. 이때 일부 서열만 지정하여 보여주거나 반지름의 크기, 선의 두께 등 일부 속성을 지정해 줄 수 있습니다.
# Karyotype 파일 포맷
chr - ID LABEL START END COLOR (chromosome의 경우)
band hs1 p36.33 p36.33 0 2300000 gneg (Cytogenetic bands의 경우)
3. 데이터의 시각화 속성을 지정해주는 configuration 파일 생성
Configuration 파일은 Circos 프로그램 실행을 위한 명령문들을 정의한 파일로써 앞서 준비한 Karyotype 파일 및 기타 data input file들을 지정해주고 각 데이터 트렉의 위치 및 크기, 색상, 테두리 선 등 다양한 시각화 방법에 대한 지침을 포함하고 있습니다. 색상이나 글꼴과 같이 쉽게 바뀌지 않는 값들은 기존에 셋팅된 configuration 파일이 있다면 <<include ...>>를 이용하여 import 후 이용할 수 있습니다.
(http://circos.ca/tutorials/lessons/configuration/configuration_files/ 참고)
링크된 페이지 (http://circos.ca/documentation/tutorials/configuration/configuration_files/images)에는 Circos가 제공하는 다양한 색상 및 글꼴 정보가 있으니 참고하시면 map의 퀄리티를 한층 업그레이드하실 수 있습니다. 물론 해당 정보를 가지고 있는 configuration 파일이 별도로 존재하므로 import해 주어야 프로그램이 인식할 수 있겠죠?
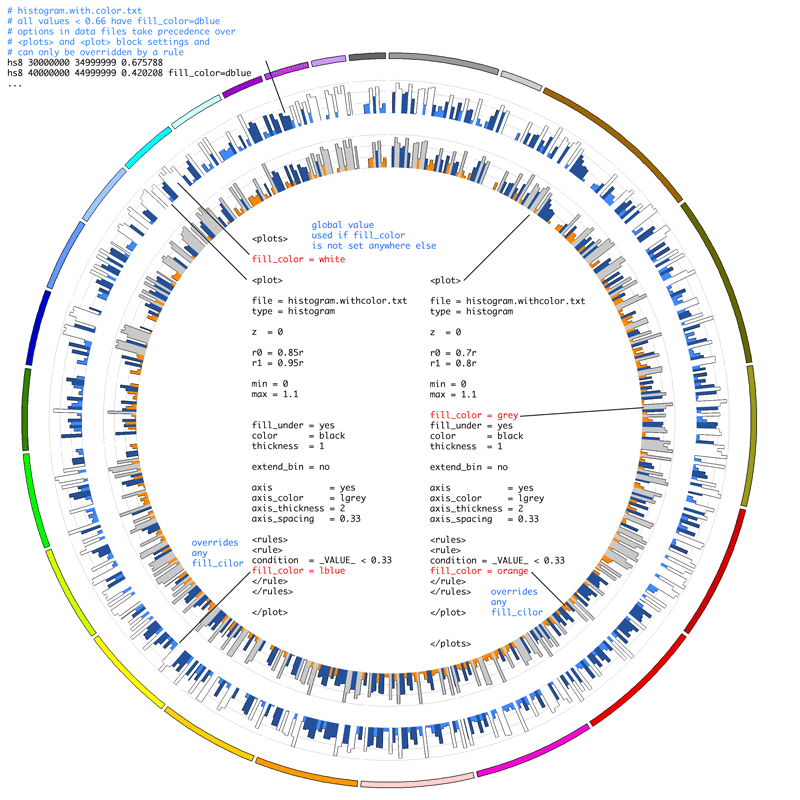
그림 3. Configuration file 예시
4. Circos 실행하기
Circos 프로그램의 실행은 아래와 같이 configuration 파일만 지정해주면 됩니다.
$ cd circos-x.xx/bin
$ ./circos -conf circos.conf
데이터를 어떻게 시각화할 것인지에 대한 깊은 고뇌와 manual을 살펴보며 만든 여러 input file들, 그리고 시키는 대로 한 것 같은데 자꾸만 에러를 뱉고 죽어버리는 야속한 configuration 파일 때문에 포기하고 싶을 수많은 순간들을 이겨낸다면 결국에는 완성된 Circos map (PNG/SVF format)을 얻을 수 있습니다. 그러나 노력의 결과와 달리 막상 완성된 Circos map을 열어 보면 색상 배열이나 plot size가 적절하지 않아 기대했던 만큼 예쁜 그림을 얻기 어렵습니다. 다시 값들을 변경하고 실행하기를 여러 차례 거친 후에야 원하는 최적의 map을 완성할 수 있을 것입니다. 최종적으로 논문화하기 위해서는 별도로 범례 등을 추가해 주어야 한다는 함정이 있습니다.
Case I. Genome visualization
[그림 4]는 약간의 표현형 차이를 보이는 10개의 샘플에 대해서 26개의 타겟 유전자 좌위를 비교한 결과 동일한 유전자를 가지고 있으면 link로 표현한 Circos map입니다. Karyotype은 10개의 genome에 대한 정보를 가지고 있으며 동일한 종이므로 genome size를 같게 표현하였습니다. 각 유전자는 tile type으로, 유전자 간 연결선은 link type으로 나타냈는데 이때 구별이 용이하도록 각각 다른 색상을 지정해 주었습니다. plot의 색상은 input file에서 추가해 주면 됩니다. 만약 유전자 별로 트렉 위치 (radius)나 크기를 다르게 표현하고자 했다면 각각의 input file을 만들어 configuration 파일에 지정해 주어야 합니다.
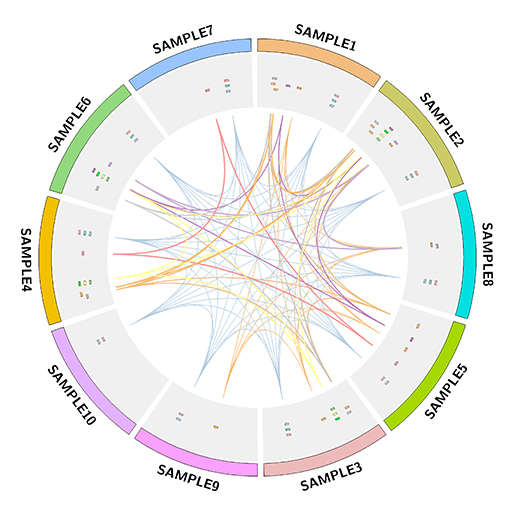
그림 4. 10개 샘플의 표현형 관련 유전자 비교 분석
이해를 돕기 위해 분석시 사용했던 input file (일부) 및 configuration file을 덧붙이자면 다음과 같습니다. external configuration file의 경우 웬만하면 아래와 같이 모두 import 후 시작하는 것이 정신 건강에 유익합니다. 이때 Circos 프로그램의 실행 위치와 각 파일들의 위치를 잘 파악하여 필요시 full path로 적어주셔야 한다는 점~ 기억해 주세요.
1. Configuration file
# External configuration file 지정하기
<<include etc/colors_fonts_patterns.conf>>
<<include etc/colors.brewer.conf>>
<<include ideogram.conf>>
<<include ticks.conf>>
<<include housekeeping.conf>>
# Karyotype 파일 지정 및 나타낼 chromosome 지정하기
karyotype = karyotype.txt
<image>
<<include etc/image.conf>>
</image>
chromosomes_units = 1000000
chromosomes_order = Sample1,Sample2,Sample8,Sample5,Sample3,Sample9,
Sample10,Sample4,Sample6,Sample7
show_ticks* = no
# etc/colors.brewer.conf 파일에 선언되어 있는 color alias를 이용하여
각 chromosome의 색 지정하기
<colors>
Sample1 = lum80chr1
Sample2 = lum80chr3
Sample3 = lum80chr7
Sample4 = lum80chr9
Sample5 = lum80chr11
Sample6 = lum80chr13
Sample7 = lum80chr15
Sample8 = lum80chr17
Sample9 = lum80chr19
Sample10 = lum80chr21
</colors>
# link type으로 유전자간 연결 선 지정하기
<links>
<link>
file = gene_link.txt
radius = 0.7r
thickness = 3
</link>
</links>
# plot type으로 각 유전자 나타내기
<plots>
<plot>
type = tile
file = gene.txt
orientation= center
label_size = 3
r0 = 0.73r
r1 = 0.99r
padding = 20p
rpadding = 20p
stroke_thickness = 1
stroke_color = black
<backgrounds>
<background>
color = vvlgrey
</background>
</backgrounds>
</plot>
</plots>
2. Karyotype file
chr - Sample1 Sample1 0 251057686 Sample1
chr - Sample1 Sample2 0 251057686 Sample2
3. gene_link.txt file
Sample1 37664116 37667348 Sample2 37664116
37667348 color=lum80chr1
Sample1 37664116 37667348 Sample5 37664116
37667348 color=lum80chr1
4. gene.txt file Sample1 32664117 42667349 color=lum80chr1 Sample2
32664117 42667349 color=lum80chr1 Sample5 32664117 42667349
color=lum80chr1
Case II. Comparative genome visualization
하나의 genome에 대해서 structural annotation 분석을 수행 후 얻어진 유전자 좌위를 표현한 것이 [그림 5]입니다. Case I과 마찬가지로 유전자는 plot type으로 지정해 주었고 유전자의 functional category에 따라서 다르게 색을 부여하였습니다. 이때 forward strand 유전자의 경우 바깥쪽에, reverse strand 유전자의 경우 안쪽에 배치하여 유전자의 방향성을 표현하였습니다. 이를 위해서 각 strand 별로 input file을 달리하여 configuration 파일에서 plot의 radius를 조정하였습니다. 그림에서 볼 수 있는 범례는 다른 툴을 이용하여 추가해 준 것으로 Circos에서 이 기능이 포함된다면 좀더 유용하게 이용할 수 있을 것 같습니다.
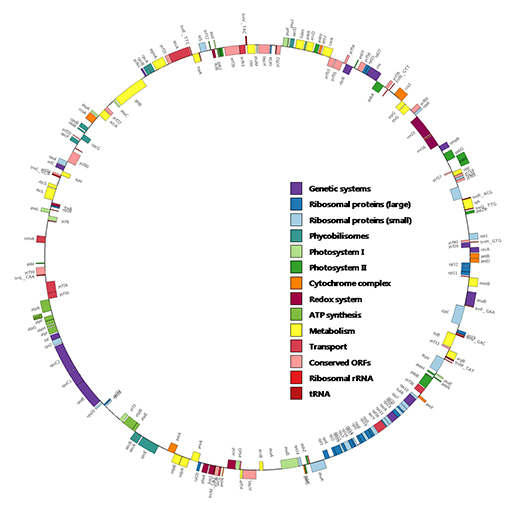
그림 5. Circos로 표현한 genome browser
작성자 : Codes사업부 Research실
정명희 컨설턴트
Posted by 人Co


 입사지원서.docx
입사지원서.docx