
[Quipu Issue Paper] Variation study Ⅲ - CNV(Copy Number Variation) Analysis
- Posted at 2010/02/18 09:17
- Filed under 생물정보
연재 순서
1. Assembly
2. Variation study
3. Expression study
4. Epigenomics
5. Genome Annotation
6. Next Generation Bioinformatics
7. Data Management for web 2.0 Era
8. Semantic Network for Integrated Biology Data
9. Gene Network Discovery by Text-mining
10. Centralization for High-throughput Data Analysis
이번 연재에서는 Next Generation Sequencing의 첫 번째 Application인 Variation study 중에 CNV (Copy Number Variation) 분석법에 대해 알아보도록 하겠습니다.
2-1-2. CNV (Copy Number Variation) Analysis
SNP가 유전적 다형성의 대명사로 여겨졌지만 이외에도 정상 표현형인 인간의 유전체에 유전자 복제 수(copy number) 변이가 존재하여 유전적 다양성에 기여하고, 암 또는 많은 질병 감수성과도 연관될 가능성이 높다는 연구 결과가 보고되면서 유전체의 구조적 변이에 대한 관심이 대두되었다. CNV(Copy Number Variants)는 reference 유전체와 비교해서 copy number의 차이를 보이는 1kb 이상의 DNA 조각으로 정의하며, 평균 크기는 29kb에서 523kb 정도로 예상된다고 한다.
현재 전체 유전체에서 CNV를 발굴하는 방식 중 가장 흔히 사용되는 방식은 CGH (comparative genomic hybridization)의 원리에 DNA 칩의 기술을 접목시킨 array-CGH이다. 마이크로어레이 기반 CGH 실험 분석 목적은 모든 유전체 안에서 각각의 유전자 조각들이 반복 횟수 변화를 보이는 부분을 선별해 내거나 반복 횟수의 양적 변화를 찾는 것이다. 이렇게 마이크로어레이 플랫폼을 이용해 발굴된 CNV는 분석에 이용된 플랫폼 의존 특성을 가지게 되어 최종 데이터의 질적인 측면과 연관되어 분석 결과의 치우침 문제를 유발할 수 있다. 또한 hybridization 효율이 프로브 마다 다양하고, 실제 copy number의 프로브 서열이 아닐 가능성도 고려해야 하는 한계에 봉착하였다. 이에 이를 극복할 만한 대안이 필요한 상황에서 NGS 기술의 보급은 CNV 발굴의 차세대 플랫폼으로 등장하였다. 앞서 언급된 NGS 기술을 통한 SNP 분석과 마찬가지로 유전체 서열과 다양한 fragment size의 paired-end reads를 assembly 함으로써 시퀀싱 coverage를 이용한 잠재적인 CNV를 분석할 수 있다(그림 4).
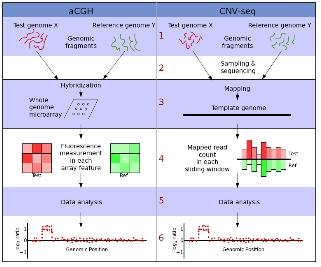 그림 4. aCGH와 CNV-seq 방법의 분석 과정 비교
그림 4. aCGH와 CNV-seq 방법의 분석 과정 비교
다음 연재에서는 전체 유전체의 염기서열 분석이 아닌 관심있는 특정 유전체의 일부분을 분석하는 방법인 Sequence Capture 기술에 대해 알아보도록 하겠습니다.
많은 관심 부탁드립니다.
참고문헌
1. Assembly
2. Variation study
3. Expression study
4. Epigenomics
5. Genome Annotation
6. Next Generation Bioinformatics
7. Data Management for web 2.0 Era
8. Semantic Network for Integrated Biology Data
9. Gene Network Discovery by Text-mining
10. Centralization for High-throughput Data Analysis
이번 연재에서는 Next Generation Sequencing의 첫 번째 Application인 Variation study 중에 CNV (Copy Number Variation) 분석법에 대해 알아보도록 하겠습니다.
2-1-2. CNV (Copy Number Variation) Analysis
SNP가 유전적 다형성의 대명사로 여겨졌지만 이외에도 정상 표현형인 인간의 유전체에 유전자 복제 수(copy number) 변이가 존재하여 유전적 다양성에 기여하고, 암 또는 많은 질병 감수성과도 연관될 가능성이 높다는 연구 결과가 보고되면서 유전체의 구조적 변이에 대한 관심이 대두되었다. CNV(Copy Number Variants)는 reference 유전체와 비교해서 copy number의 차이를 보이는 1kb 이상의 DNA 조각으로 정의하며, 평균 크기는 29kb에서 523kb 정도로 예상된다고 한다.
현재 전체 유전체에서 CNV를 발굴하는 방식 중 가장 흔히 사용되는 방식은 CGH (comparative genomic hybridization)의 원리에 DNA 칩의 기술을 접목시킨 array-CGH이다. 마이크로어레이 기반 CGH 실험 분석 목적은 모든 유전체 안에서 각각의 유전자 조각들이 반복 횟수 변화를 보이는 부분을 선별해 내거나 반복 횟수의 양적 변화를 찾는 것이다. 이렇게 마이크로어레이 플랫폼을 이용해 발굴된 CNV는 분석에 이용된 플랫폼 의존 특성을 가지게 되어 최종 데이터의 질적인 측면과 연관되어 분석 결과의 치우침 문제를 유발할 수 있다. 또한 hybridization 효율이 프로브 마다 다양하고, 실제 copy number의 프로브 서열이 아닐 가능성도 고려해야 하는 한계에 봉착하였다. 이에 이를 극복할 만한 대안이 필요한 상황에서 NGS 기술의 보급은 CNV 발굴의 차세대 플랫폼으로 등장하였다. 앞서 언급된 NGS 기술을 통한 SNP 분석과 마찬가지로 유전체 서열과 다양한 fragment size의 paired-end reads를 assembly 함으로써 시퀀싱 coverage를 이용한 잠재적인 CNV를 분석할 수 있다(그림 4).
그러나 SNP와 같이 하나의 염기서열 차이로 변이를 확인하는 것이 아니기 때문에 assembly 분석 시 시퀀싱 오류로 인하여 다른 부분에 정렬되어 잘못된 variation을 검출하게 되는 가능성도 배제할 수는 없다. 따라서 최근 Robust 통계 모델을 기본으로 하면서 aCGH와 NGS 기술의 이점들만 조합하여 효율적인 CNV 분석에 대한 논문이 발표되었고 이러한 방법을 이용하여 두 개체(Dr. J. Craig Venter와 Dr. James Watson) 사이의 CNV를 분석한 평가 결과도 함께 확인할 수 있어 이 후 aCGH와 NGS 기술을 접목한 CNV 분석 방법이 충분히 발전할 것으로 생각된다[4]. 이렇게 진행한 연구 방법과 결과들은 웹사이트를 통하여 무료로 이용할 수 있다(http://tiger.dbs.nus.edu.sg/CNV-seq).
다음 연재에서는 전체 유전체의 염기서열 분석이 아닌 관심있는 특정 유전체의 일부분을 분석하는 방법인 Sequence Capture 기술에 대해 알아보도록 하겠습니다.
많은 관심 부탁드립니다.
참고문헌
1. 이종극 (2006) 질병유전체분석법(Genetic Variation and Diseases)
2. Eck SH, Benet-Pagès A, Flisikowski K, Meitinger T, Fries R, Strom TM. (2009) Whole genome sequencing of a single Bos taurus animal for single nucleotide polymorphism discovery. Genome Biol. 10(8), R82.
3. Ganal MW, Altmann T, Röder MS. (2009) SNP identification in crop plants. Curr Opin Plant Biol. 2, 211-217
4. Xie C, Tammi MT. (2009) CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 10, 80
5. Illumina : SNP Genotyping and CNV Analysis
(http://www.illumina.com/documents/products/datasheets/datasheet_genomic_sequence.pdf)
6. Bentley DR. et al. (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008 456, 53-59
7. Ng SB. et al. (2009) Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 461, 272-276
8. Koboldt DC, Miller RD, Kwok PY. (2006) Distribution of human SNPs and its effect on high-throughput genotyping. Hum Mutat. 3, 249-254.
9. 박종화 (2009) 변이체학을 위한 생정보학 분석도구. Medical POSTGRADUATES. 3(37), 131-133
10. 유향숙, 김선영 (2009) Variome 국제연구동향. Medical POSTGRADUATES. 3(37), 134-137
11. 임선희, 정연준. (2009) 새로운 유전체 변이의 등장 : 유전자 복제수 변이. Medical POSTGRADUATES. 3(37), 149-1532. Eck SH, Benet-Pagès A, Flisikowski K, Meitinger T, Fries R, Strom TM. (2009) Whole genome sequencing of a single Bos taurus animal for single nucleotide polymorphism discovery. Genome Biol. 10(8), R82.
3. Ganal MW, Altmann T, Röder MS. (2009) SNP identification in crop plants. Curr Opin Plant Biol. 2, 211-217
4. Xie C, Tammi MT. (2009) CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 10, 80
5. Illumina : SNP Genotyping and CNV Analysis
(http://www.illumina.com/documents/products/datasheets/datasheet_genomic_sequence.pdf)
6. Bentley DR. et al. (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008 456, 53-59
7. Ng SB. et al. (2009) Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 461, 272-276
8. Koboldt DC, Miller RD, Kwok PY. (2006) Distribution of human SNPs and its effect on high-throughput genotyping. Hum Mutat. 3, 249-254.
9. 박종화 (2009) 변이체학을 위한 생정보학 분석도구. Medical POSTGRADUATES. 3(37), 131-133
10. 유향숙, 김선영 (2009) Variome 국제연구동향. Medical POSTGRADUATES. 3(37), 134-137
Posted by 人Co
- Tag
- aCGH, Assembly, CNV, NGS, SNP, variation, 마이크로어레이, 생물정보분석
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/42
Trackback URL : 이 글에는 트랙백을 보낼 수 없습니다

