
ISMB 2014 참석 후기
- Posted at 2014/09/03 17:54
- Filed under 생물정보
어느덧 22번째 ISMB2014
하버드와 MIT로 유명한 미국의 대표적인 교육 도시, 랍스터의 천국, 미국 보스턴에서 생물정보학자들의 큰 축제, ISMB(International Conference on Intelligent Systems for Molecular Biology)가 진행되었습니다. 이번 ISMB2014는 어느덧 22번째를 맞이했습니다. (세계에서 가장 큰 생물정보학/전산생물학 관련 학회)

ISMB는 Intelligent Systems for Molecular Biology 라는 주제로 매년 개최되고 있습니다.
2014년 7월 11일 부터 15일까지 4박 5일 동안 열린 이번 ISMB2014는 미국 보스턴의 John B. Hynes
Memorial Convention Center 에서 진행되었습니다. ISMB는 ISCB (International Society
for Computational Biology)에서 주최하는데, 70개의 나라에서 모인 3,000 명 이상의 멤버들이 꾸려나가는
모임으로 바이오인포메틱스(Bioinformatics) 저널과 PLoS(Public Library of Science) 라는 오픈
엑세스 저널을 통해 편찬 작업을 진행하고 각종 미팅과 컨퍼런스 교육 등 다양한 활동을 합니다. 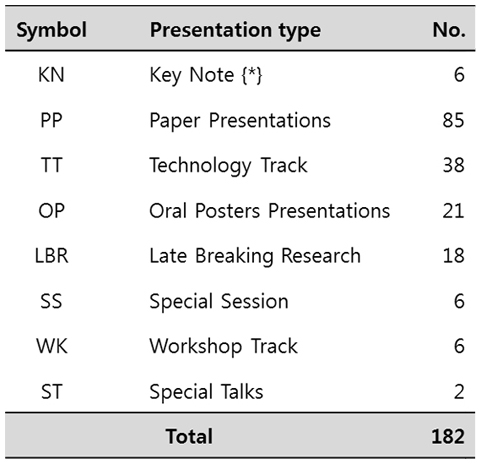
총 5일 동안 진행된 이번 학회는 11~12일 이틀 동안은 교육자, 학생을 위한 프리-컨퍼런스,
13~15일까지는 메인 컨퍼런스로 구성되었습니다. 메인 컨퍼런스는 총 8개의 프리젠테이션 타입이 있고, 키노트를 제외한 나머지
7개 타입이 동시다발적으로 진행되었습니다. 키노트는 ISCB 멤버중 당해 년도에 가장 업적이 좋은 6분이 나와서 발표를 하였고,
페이퍼 프리젠테이션은 ISCB를 통해 발표된 논문 중 선별된 내용이었습니다. 이번 학회의 주요 주제는 다음의 내용과 같습니다.
- Sequence analysis
- Comparative genomics
- Gene regulation and transcriptomics
- Systems biology
- Databases and data integration
- Text mining and information extraction
- Human health

"생명은 유전자가 아니라 정보의 흐름으로 이해해야 한다"
두번째로는, 이쪽 생물정보 분야의 연예인인 Eugene Myers 입니다. 서열을 다루는 생물학자라면 누구나 한번은 사용해봤을 법한 NCBI의 BLAST가 이분을 통해서 만들어졌습니다. 그것만으로도 대단한데 Human Genome Project 당시 셀레라 지노믹스의 샷건 시퀀싱 기술을 Eugene Myers 가 만들었다고 합니다. 물론 다국적팀이 3일 먼저 완료했다는 보고 덕에 과학계에서는 큰 의미없는 아쉬운 2등을 했지만, 굉장히 짧은 시간에 지놈을 완성할 수 있는 알고리즘을 개발했다는 측면에서 다국적팀의 Jim Kent 보다 더 유명해지지 않았나 싶습니다. DNA assembly 라는 주제로 긴 시간동안 많은 고견을 들을 수 있었습니다. 앞으로는 low fold 의 시퀀싱을 가지고 어셈블리를 할 것이고, PacBio 같은 long read 를 가지고 퀄리티가 좋은 어셈블리를 하는 시대를 예견했습니다. Long reads 를 준비하는 이유는 먼 훗날 언젠간 어셈블리가 필요 없어질 것이라는 이야기도 했는데 PacBio에서 이번에 새로 출시되는 시퀀서에서는 한방에 complete transcripts가 시퀀싱 된다고 합니다. 현재의 난제인 지놈도 언젠간 가능하지 않을까라는 생각을 해봅니다.
더불어 생물정보학의 중요성을 다시 한 번 깨닫게 해 주었던 키노트 중 하나는 스탠포드 대학교의 Russ Altman 박사가 발표한 " Informatics for understanding drug response at all scales " 입니다. 그는 분자, 집단, 종 등 다양한 drug response 에 대해서 발표했습니다. 약에 대한 연구에서 가장 중요한 것 중 한 가지는 다양한 레벨에서의 반응을 이해해야 한다는 것입니다. 한두 가지 특정 상황이 아니라 성질, 반응, 상호 작용, 구조, 유전자와의 상호 반응, 3차 구조 등 모든 다양한 분야에서의 정보 융합이 필요하다고 제창하였습니다. 즉, 가용할 수 있는 모든 정보가 개인 맞춤의학에 대해서 도움을 줄 수 있으며, 이제 "생명은 유전자가 아니라 정보의 흐름으로 이해해야 한다" 라는 큰 틀을 이야기했습니다.
- Good things come in Small Packages – Replicators and Innovators – Michal Linial
- DNA Assembly : Past, Present, and Future – Eugene(Gene) Myers
- Biomedical Quants of the world Unite! We only have our disease burden to lose – Isaac(Zak) Kohane
- Biomaterials and biotechnolgy: From the discovery of the first angiogenesis inhibitors to the development of controlled drug delivery systems and the foundation of tissue engineering – Rober Langer
- Informatics for understanding drug response at all scales – Russ Altman
- Multidimensional single cell approach to understand cellular behavior – Dana Pe’er
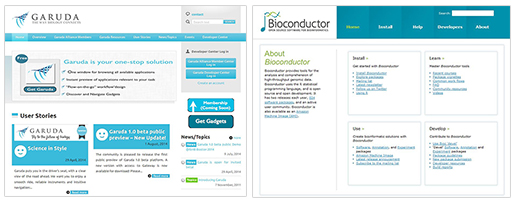
생물학자들을 위한 생물정보 어플리케이션
또한, 기술적인 부분에서는 생물학을 연구하는 다양한 학자들을 위하여 많은 어플리케이션이 소개되었습니다. 일본의 리켄에서 제공하는 가루다 시스템과 NGS 분석에서 한 번쯤은 사용해 본 적이 있는 Bioconductor 패키지에 대한 자세한 설명과 사용법에 대해서도 소개하는 세션이 있었습니다. BioConductor에서는 800여개가 넘는 다양한 분석 모듈을 제공함으로써 손쉽게 생물데이터를 다룰 수 있도록 했고 교육, 메뉴얼, 동영상 등 초보자도 쉽게 따라서 할 수 있도록 많은 정보를 제공하고 있다는 것을 소개해주었습니다. 그리고 웹에서 NGS 데이터를 분석할 수 있는 환경을 만들어 주는 Galaxy에서는 기본 설치에서부터 사용법, API 등의 사용법 다양한 정보를 제공해주었습니다. 특히 그들 그룹에서는 현재 제공되는 Galaxy 뿐 만 아니라 다양한 주요 분석 패키지를 취합해 하나의 어플리케이션으로 제공할 계획을 세우고 있다고 합니다. 그런 부분에서 그들의 도전 정신을 깊이 배울 수 있는 계기가 되었습니다.
메인컨퍼런스의 한 꼭지인 포스터 발표장입니다. 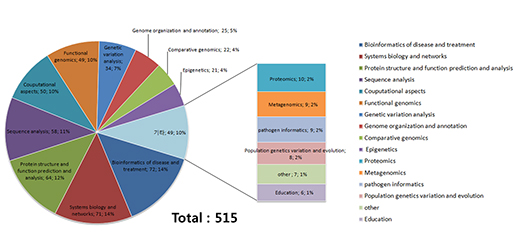
Human & Disease & Bioinformatics
각 분야에서도 특히 human 쪽의 연구가 많이 진행되고 있는 것을 확인할 수 있었습니다. 이에 발맞춰 human에 특화된 RNA-seq normalization 방법을 소개한 포스터도 있었습니다. 펜실베니아 대학의 한 연구팀은 서로 다른 샘플간 같은 feature에 대한 비교에 사용된 기존의 normalization 방법의 8가지 문제점을 나열하고 이를 반영한 새로운 normalization 방법을 고안했습니다. 대부분의 문제점은 분석 목적에 맞는 RNA-seq normalization 방법에 분명 필요한 것들이고, 당사의 RNA-seq normalization 방법에도 반영되고 있는 부분이었습니다. 하지만 한가지 눈길을 끄는 새로운 방법이 눈에 띄었습니다. 앞서 말씀드린 대로 다른 샘플간의 같은 feature를 보고자 한다면 그 비교의 범위를 좀 더 명확하게 볼 필요가 있습니다. 이를 위해서 dominant gene 을 제거합니다. 이러한 gene들은 house keeping gene 이나 special case gene 을 말하는데 이와 같은 very high expressed gene 을 제거함으로써 보고자 하는 대상을 좀 더 세밀하게 관찰할 수 있도록 합니다. 또한, transcript 단위가 아닌 exon 단위별 mapped reads 개수를 categorization 하여 RPKM 편차를 줄이는 방법을 사용하는데 이를 통해 좀 더 유의한 비교가 가능하다고 설명합니다. 이런 방법들은 연구의 목적에 따라 개연성에 맞게 적용되어야 하는 부분이지만 새로운 normalization 기법을 생각해 볼 수 있었습니다.

미국에서 개최된 학회라서 그런지 많은 연구자의 소속이 미국소재지였습니다. 물론, 다양한 국가출신의 연구자들이 함께한 학회였지만,
그들이 자리한 자유와 기회의 땅으로 불리는 미국의 기운을 받아 자유로운 생각과 창의적인 아이디어로 연구를 진행하고 리드한다는
느낌을 받았습니다. 미국문화의 내면과 외면에는 많은 차이가 있겠지만, 이런 공부와 연구에 대한 환경을 제공하는 시스템은 우리가
본받을 점이라고 생각합니다. 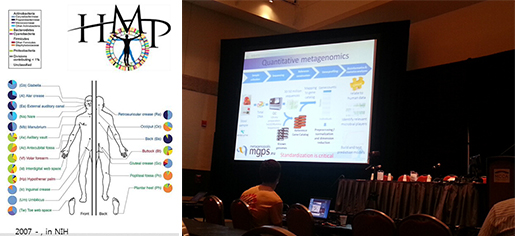
이번 ISMB에서는 학회 주제에 맞는 다양한 주제들을 접할 수 있었습니다. 특히 휴먼에 대한 질병과 치료연구들이 많이 진행되고 있었고 BIOBASE와 EMBL 쪽에서도 cancer 연구에 특화된 데이터베이스를 준비하고 있었습니다. 한편 제 2의 genome이라 불리는 microbiome 연구 또한 탄탄한 펀드를 기반으로 활발히 연구되고 있습니다. NIH에서 2007년부터 시작된 HMP (Human Microbiome Project)뿐만이 아니라 유럽에서는 개개인의 microbiome 을 이용한 치료를 목적으로 5,000명의 데이터베이스를 준비하고 있다고 합니다. Microbiome 연구는 앞으로 전 세계적으로 확장될 분야라는 확신을 얻었고 앞으로 국내에도 많은 연구결과 소식을 접할 수 있기를 기대합니다.
작성자 : Codes 사업본부 Development팀 이규열 팀장,
Research실 유승일 컨설턴트
Posted by 人Co
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/160
Trackback URL : 이 글에는 트랙백을 보낼 수 없습니다

