
NGS 데이터로부터 유전체 크기를 예측하는 방법
- Posted at 2013/06/11 16:27
- Filed under 생물정보
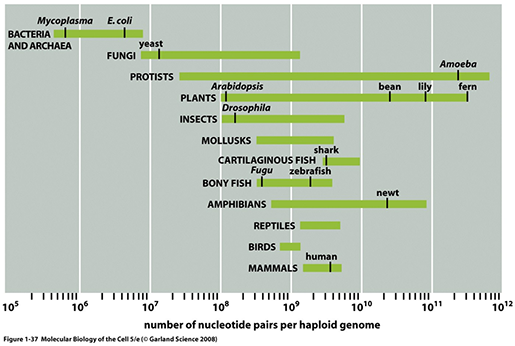
Genome size의 다양성
위 그림에 보이는 것처럼 생물종군에 따라서 유전체 사이즈는 상당히 넓은 범위에 걸쳐있는 것을 알
수 있습니다. 주목할 것은 흔히 생각하듯이 그 생물체의 organismal complexity와 유전체 크기는 큰 상관관계가
없을 수도 있다는 것입니다. 예를 들어 human보다 수십배, 수백배 더 큰 유전체 크기를 자랑하는 Protist, Plant,
Amphibian 종들도 존재하는것을 확인할 수 있습니다 ("C value paradox"). 생물종의 복잡도와 유전자 개수,
그리고 유전체 크기등의 상관분석도 재미난 연구 topic이 될 수 있을 것 같고 아마 누군가는 열심히 연구 중에 있을 것 같습니다
(아마도 결과가 나와있을지도??).
생물정보학적인 유전체 분석의 제일 첫 스텝은 아무래도 de novo genome assembly가 아닐까 합니다. 만
약에 어떤 생물종의 유전체 de novo assembly를 진행하고자 할때 사전지식이 없어 분석하고자 하는 그 생물종의 유전체
크기를 알지 못한 상태에서 진행한다면 어떨까요? 염색체가 몇개인지 전체 사이즈가 얼마인지 모른다면 상당히 답답할 것입니다. 시퀀싱을 얼마나 어떻게 해야할지도 막막할테구요. 일
반적으로는 실험적인 방법 (일례로 flow cytometry)을 통해서든 아니면 유전체 크기가 알려진 유사성이 높은 다른
종으로부터 어느정도 사이즈를 예상한 상태에서 NGS 분석을 진행합니다 (실험적인 유전체 사이즈 측정방법들에 대해서는 다음 기회에
다뤄보도록 하겠습니다). 하지만 de novo assembly를 통해 만들어낸 draft genome이 맞게 잘 만들어진 것인지 확인하기 위해서라도 대략적인 유전체 크기를 알면 많은 도움이 될 것입니다.
첫번째: BGI 방법
첫번째는 BGI (Beijing Genome Institute)에서 소개한 방법입니다. 이미 여러편의 논문에서 인용이 되었고 저희팀에서 자주 이용하는 방법입니다. 원리는 간단합니다. Genome shotgun NGS를 통해서 100bp 짜리 short reads 3400만개를 얻었다면 총 total base number는 3.4 x 10^9이 됩니다. 만약 그 종의 유전체사이즈가 200Mb라면 이는 17 fold에 해당하는 양입니다.
만약 genomic sequencing library를 유전체 조각의 유실없이 제대로 잘 만들었고 시퀀싱도 충분한 양 (15-fold 이상)이 되어졌다면 특정 시퀀스들을 대상으로 depth frequency를 plotting 했을때 17-fold에서 peak를 그리는 정규분포의 모양을 할 것이라고 예상할 수 있습니다.-- 수식 (1)
위 수식을 보면, total_read_num, avg_read_length는 이미 알고 있으므로 coverage_depth를 알면 미지의 genome size를 계산할 수 있게 됩니다.
여기서 특정 사이즈의 k-mer시퀀스를 살펴봅시다. NGS data로부터 만들어질수 있는 전체 k-mer 갯수는 다음과 같습니다.예를 들어 100-mer짜리 Illumina HiSeq read 하나에서 만들어질 수 있는 17-mer (임의의 k-mer를 17로 정했을경우)의 갯수는, 100 - 17 + 1 (= 84)개가 될 것입니다. 이러한 HiSeq reads가 300백만개가 있다면, average read length는 100-mer, k-mer size는 17-mer로 상정할 경우,
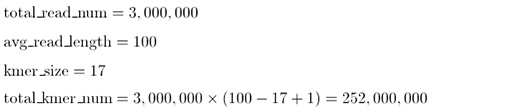
이 됩니다. 이 2억5천2백만개의 k-mer들 중에는 동일한 시퀀스들을 가진것들이 있을 것이고 unique한 k-mer들의 리스트를 작성, 각 k-mer가 몇번씩 관찰되었는지 그 frequency를 계산합니다. 그렇게 해서 depth가 같은 k-mer들이 전체 k-mer pool에서 차지하는 비율을 plotting 합니다 (x축은 k-mer depth, y축은 전체에서 차지하는 frequency, k-mer frequency plot의 example은 아래 giant panda 데이터를 참조).
그리고 다음의 수식도 성립합니다.위에서 작성한 k-mer frequency plot에서 peak을 보이는 depth point가 k-mer_coverage_depth가 되며 아래 수식에 의해 NGS 시퀀스 전체의 genome coverage depth를 구할 수 있게 됩니다.
여기서 구해진 coverage_depth로부터 전체 genome_size를 위 수식 (1)로 부터 구할 수 있습니다.
-- 수식 (2)
JELLYFISH: k-mer frequency distribution 측정
다음으로 NGS data로부터 k-mer frequency plot을 얻는 방법입니다. 여러개의 프로그램이 논문에 나와있는데 그중에서 저희팀은 BGI와 마찬가지로 JELLYFISH를 이용합니다.

Ref. Marcais and Kingsford (2011) A fast lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27:764–770
JELLYFISH는 free software로 여기(http://www.cbcb.umd.edu/software/jellyfish/)에서 다운로드 받을 수 있습니다.
JELLYFISH를 이용해 얻은 k-mer frequency plot을 얻은 일례입니다. BGI에서 분석한 Giant Panda의 경우인데, 17-mer짜리를 이용해서 frequency plot을 얻은 결과입니다.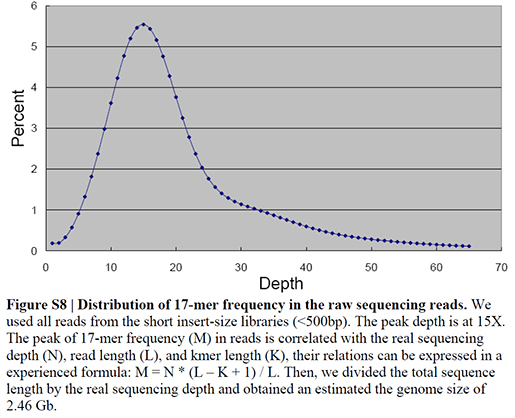
Ref. Li et al, The sequence and de novo assembly of the giant panda genome. Nat. Genet. (2010) 473:311-317
왜 17-mer인가?
이론적으로는 어떤 k-mer를 사용하든 유사한 결과를 얻을 것입니다. 하지만 제대로된 결과를 얻기위한 가정은 최소한 특정 k-mer가 해당 유전체에서 딱 한번 unique하게 발견되어야 합니다. 5-mer 짜리는 확률적으로 1 kb당 한번씩 발견될 수 있고, 10-mer는 1 Mb당 한번씩 발견될 수 있습니다. 이렇게 사이즈가 작은 k-mer들은 시퀀스가 유전체에 여러번 무작위적으로 발견될수 있기 때문에 올바른 결과를 담보할 수 없습니다. 16-mer쯤 되면 4.3 Gb당 한번정도 무작위적으로 발견됩니다. 이정도면 나쁘진 않지만 위 표에서 보시다시피 4.3 Gb 보다 더 큰 사이즈의 유전체도 존재하기 때문에 안심할 수 없겠죠. 17-mer 정도면 (17 Gb당 1번) 현재 진행되는 유전체 프로젝트에서는 안전한 결과를 보장할 수 있다고 하겠습니다. 더 큰사이즈의 k-mer는 안되냐구요? 물론 가능합니다. 하지만 frequency plot을 그리기 위해 더 많은 k-mer를 이용하는 것이 일반적으로 더 안정적인 결과를 얻을 수 있고 또 시퀀스의 base quality가 완벽하지 않기 때문에 시퀀스 에러가 있을 수 있어 큰 k-mer를 사용할 수록 에러율이 높아질 위험이 있습니다. 대략 17~21-mer 정도가 가장 효율적이라 여겨집니다.
두번째: RNA-seq 방법
두번째로 소개할 방법은 BMC Genomics (2011) 논문에 소개된 내용입니다. 버블콘이라 불리는 바다달팽이 (Conus bullatus)의 전사체를 분석한 논문인데요 (Ref. Hu et al. BMC Genomics 2011, 12:60). 기본적인 원리는 위에서 소개한 BGI 방법과 유사합니다. 차이점은 Genome Shotgun NGS data만 이용하는 것이 아니고 RNA-seq data도 함께 이용한다는 것입니다. RNA-seq에서 얻은 sequence reads에 genome NGS reads를 매핑하는 것이죠. Transcriptome을 레퍼런스로 삼아서 genome sequence reads를 붙이고 각 transcript당 평균적으로 몇 fold로 붙었는지 계산해보면 그 genome NGS reads가 genome의 몇 fold를 반영하고 있는지를 판단할 수 있다는 원리입니다. 이 방법의 장점은 genome NGS coverage가 1.5 fold 정도여도 좋은 예측결과를 보여준다는 것입니다.
이 방법을 검증하기 위해 논문에서는 C. elegans와 D. melanogaster의 NGS 데이터로부터 가상의 1.5 fold 짜리 모의데이터를 구성해서 테스트를 해보았는데 다음과 같은 좋은 결과치를 얻을 수 있었다고 합니다.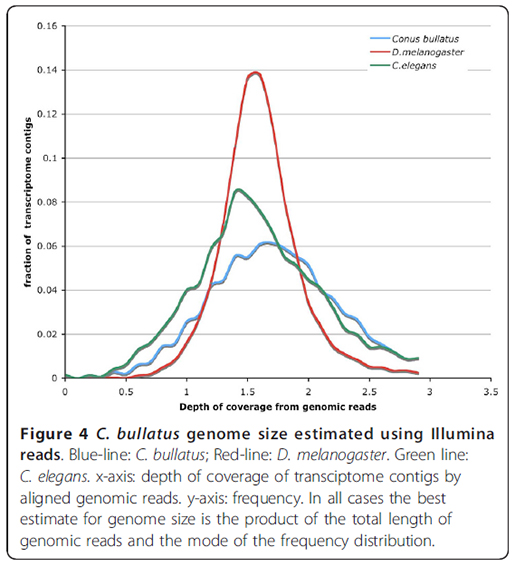
WU-BLASTN으로 "M = 1 N = -3 Q =3 R = 1 wordmask seg lcmask" 옵션을 사용해서 NGS reads를 RNA-seq 어셈블리로 만들어진 transcript contigs에 매핑합니다. 각 transcript별 coverage depth를 length 기준으로 계산해서 얻은 다음, 위 그래프에서 보는 것처럼 coverage-depth vs transcript percentage plot을 얻습니다. 이때 얻어진 maximum peak coverage가 전체 genomic NGS reads의 coverage가 되며, 수식 (2)를 이용해서 전체 genome size를 예측할 수 있습니다. 만약 소량의 genome NGS data만 있어서 첫번째로 소개한 k-mer 방법을 사용하기 어려운 상황에서 RNA-seq data를 같이 가지고 있다면, 써볼만한 방법이라고 생각됩니다.
Posted by 人Co
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/133
Trackback URL : 이 글에는 트랙백을 보낼 수 없습니다

