Figure 3. With Qiagen
Trend and methodsISMB에서는 현재...
최신분석기법 및 도구
ABySS로 유명한 GSC에서 이번 ISMB/ECCB2015를 통해 새로운 scaffolding tool인 LINKS를 소개했다. LINKS는 Long Interval Nucleotide K-mer Scaffolder의 약자로 Oxford의 Nanopore Technologies Ltd. 등을 통해 얻을 수 있는 long reads를 이용해 scaffolding한다. 이는 scaffolding이나 re-scaffolding을 수행하기 위한 새로운 방식의 유전체 조립도구다.
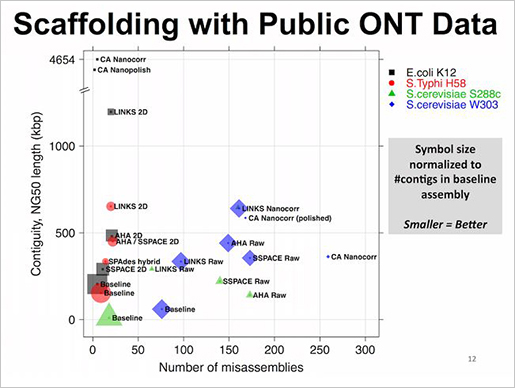
현재까지 공개된 scaffolding 도구들과 비교를 했을 때도 mis-assemblies가 적고 contiguity나 NG50길이가 긴 것을 확인 할 수 있다.
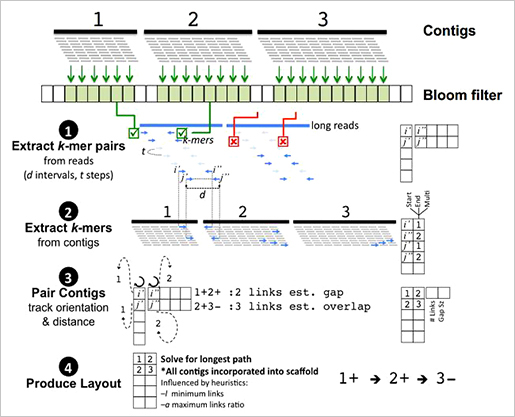
LINKS는 메모리 효율성이 매우 뛰어나다. 그 이유는 scaffolding algorithm에 있다. long reads를 짧은 k-mer pairs로 추출하고 scaffolding의 대상이 되는 contigs도 k-mer pairs를 추출한다. 각각에서 추출된 k-mer pairs의 서열상동성이 같은 위치정보와 paired-end information을 통해 scaffolding을 수행한다. 또한 iteration 수를 높게 조절함으로써 정확도를 향상할 수 있다는 장점도 존재한다.
BactoGeNIE는 NGS의 application인 comparative genome analysis를 large-scale로 수행하는 도구다. 단순한 visualization일지 모르는 이 도구는 ‘comparative gene neighborhood analysis'를 모토로 개발됐다.

위 사진은 E.coil의 700 strains에 대해서 neighborhood around a hypothetical protein을 확인하는 장면이다. Display는 at 21.9 by 6.6 feet and 6144 by 2304 pixcels이다.
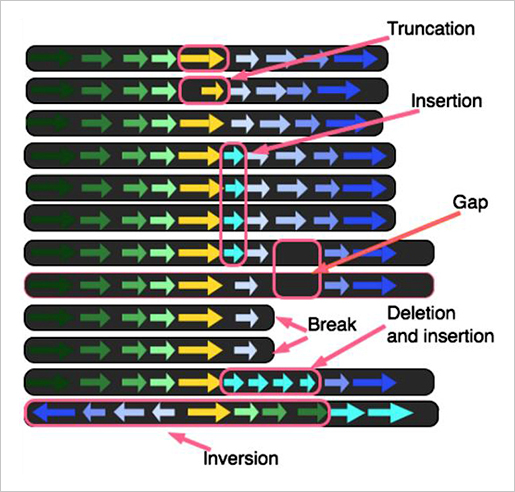
각각의 genome은 한 행에 하나씩 배열되며 배열된 모든 genome의 alignment를 통해 comparative genome analysis를 수행한다. 각각 유전체의 특징적 단위가 화살표로 표시되며 breaks, deletions, insertions, gaps 등을 확인 할 수 있다.
1988년 첫 Clustal이 소개되고 나서 1994년 ClustalW, 1997년 ClustalX, 2007년 ClustalW2에 이르기까지 Clustal 시리즈는 Multiple Sequence Alignment(MSA) Bioinformatic tools로써 많은 사랑을 받아왔다. 이번 ISMB/ECCB2015에서는 Clustal Omega의 새 버전을 소개했다.

Clustal Omega는 최초 guide-trees를 생성하기 위해 mBed (Blackshields, 2010) calculates distance matrix를 채택했다. 이로써 기존에 large (N > 10,000) alignments distance matrix가 갖는 bottleneck을 해결했다. Fabian Sievers(University College Dublin)의 말에 따르면, Clustal 시리즈의 고질적 문제였던 ‘any size'의 alignment가 가능해졌고, 퍼포먼스 또한 크게 좋아졌다.
데이터베이스
UniProt은 protein sequence와 그 functional information을 담고 있는 데이터베이스로 bioinformatics 연구에서 없어서는 안 될 존재가 됐다. UniProt을 관리하고 있는 EMBL-EBI는 최근 UniProt 데이터베이스의 대용량화에 대비해 “Proteome redundancy reduction" 프로젝트를 진행했다. 실제로 데이터베이스가 방대해짐에 따라 연구자의 컴퓨팅 환경조건이 높아지는 실정에 대해 불만의 목소리가 많았다. 이 프로젝트를 통해 92 million (2015_03 release)에서 46 million (2015_04 release)으로 감소했다.

데이터의 내용을 보호하면서 사이즈만을 어떻게 줄일 것이냐? 라는 질문에 가장 중요한 단계는 sequence comparison이라 답했다. CD-Hit-2D를 사용해 두 세트의 서열을 비교했으며 90%이상의 sequence identity threshold와 90%이상의 proteome similarity threshold로 서열상 redundancy를 제거했다.
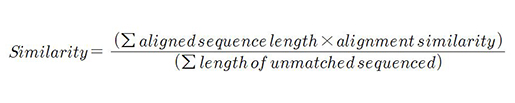
매우 많은 entry가 줄어서 데이터베이스의 크기 자체는 줄었지만 훨씬 specific하고 meaningful한 데이터베이스가 탄생했다. 줄어든 데이터베이스는 ordering을 통해 여러 개의 component로 merge되어있다. 현재 UniProt site에 released database는 proteome redundancy removal 버전이다.
EVA는 European Variation Archive의 약자로 EMBL-EBI에서 만든 genetic variation resource이다. EVA의 지향점은 모든 타입의 종과 분포를 반영하는 변이를 담는 것이 목적으로, germline은 물론 cancer genomes의 변이 또한 포함하는 것이다. 최근까지의 EVA는 13종에서 4억개의 unique variation을 담은 1TB의 데이터를 제공하고 있다. Open-access database로 서비스 중이며, variation browser를 통해 knowledge-base search가 가능하다.
 주요 관심사 및 최신동향
주요 관심사 및 최신동향
- Single cell RNAseq studies
일반적인 RNAseq profiles은 약 100,000개 이상 세포 풀링(pooling)을 통해 얻는다. 이번 연구에서는 single cell RNA-sequencing technologies를 사용하여 single cell들 간에 RNA abundance 차이를 확인했다. Single cell analysis의 다양한 applications 중 novel variation studies는 cell type composition, differentiation에 적합하며, additional (confounding) expression heterogeneity는 cell cycle, apoptosis를 분석함에 적합하다.
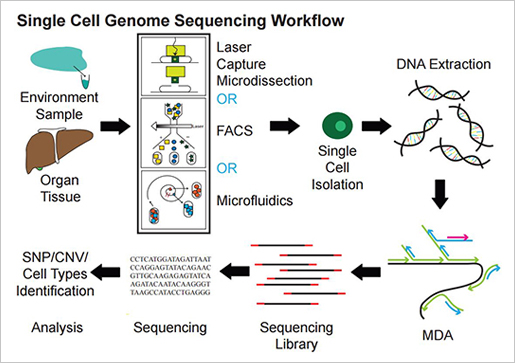
국내 연구동향 및 시퀀싱 회사의 single cell analysis는 아직 걸음마 단계에 불과하다. Single cell에서 DNA와 RNA를 분리해 시퀀싱하고 그 안에서 일어나는 생물학적 이벤트는 epigenomics 연구는 물론, cell cycle에 따른 cell-to-cell correlations, T cell 연구 등 다양한 분석에 적용이 가능하다.
Genotyping By Sequencing (GBS)는 차세대 시퀀싱 기술을 바탕으로 새롭게 개발, 발전하고 있는 NGS 분석법 중에 하나이다. 유전체 전체를 시퀀싱하는 WGS에 비해 저렴한 비용으로 빠르고 쉽게 genome-wide 분석을 가능하게 한 테크닉이다. 제한효소를 처리하여 유전체 서열에서 그 제한효소에 의해 잘리는 영역 주변의 서열만을 시퀀싱하게 된다. 제한효소를 처리한다는 점에서는 RAD-seq과 근본적으로 원리가 같지만 효소절단 후 사이즈 선별을 하지 않는다는 점에서 시퀀싱 라이브러리 제작이 보다 간단한 편이며 GBS 시퀀싱 데이터가 RAD-seq에 비해 low coverage로 얻어진다.
GBS는 아래와 같은 applications이 있다.
- Marker discovery
- Phylogeny/Kinship
- Linkage mapping of QTL in a biparental cross
- Fine mapping QTL (Quantitative trait loci)
- Genomic selection
- GWAS (Genome wide association study)
- NAM-GWAS (Nested association mapping GWAS)
- Improving reference genome assembly
GBS는 이번 ISMB2015에서도 소개됐으며, 포스터 중 눈에 띄는 분석법으로 많은 관심을 받았다. 기존 GBS 분석법/도구로 알려진 Stacks과 TASSEL을 이용하지 않고, Bowtie와 GATK를 연계한 분석법을 소개했다.
 Impression
Impression
이젠 국내에서도 bioinformatics의 인식이 달라져야 한다.
작년 ISMB2014에서는 de novo assembly 나 expression analysis, GO, KEGG, COG, pathway 연구와 같은 전반적이고 일반적인 연구가 상당수를 차지했다. 하지만 올해의 ISMB학회는 ECCB와 함께했기 때문인지 그런 비율이 많이 줄었고 좀 더 세분되고 집중적인 연구가 주를 이뤘다. 특히 암과 같은 특정 질병의 원인 판별과 치료법 개발에 초점이 맞춰져 있었다. 세션들의 주제가 GENES, PROTEINS, SYSTEMS, DATA 이외에 DESEASE도 있다는 것은 이를 반증하고 있으며, DESEASE 세션의 개수도 상당하고 그 연구수준도 점진적으로 발전되고 있어 보인다. 특히 연구방법들이 한가지 방향으로 모이지 않고 다양한 시도를 통한 기초연구에 힘쓰는 것에 놀랐다. 이는 한국의 연구풍토와 유럽, 미국의 연구풍토가 많이 다르기 때문으로 생각한다. 언제쯤인지 퇴근길에 읽은 어느 연구자님의 문구에 따르면, 정확한 메커니즘의 이해보다는 응용성을 강조하는 우리나라의 스타일과는 다르게 유럽이나 미국은 정확한 원리를 이해하고 그것을 수학적인 도구를 통해 분석해야 하며 실험의 결과들이 높은 재현성을 나타냄과 더불어 각각의 결과들이 강한 유기성을 가질 때 비로소 올바른 결과로써 인정한다고 한다. "블루오션은 찾아내는 것이 아닌 만드는 것이다.", "소비자는 자신이 무엇을 원하는지 모른다."는 명언들이 말하는 창조적 사고의 연구풍토가 선진국이 될 한국에도 자리 잡을 때가 아닌가 생각한다.
NGS의 도입과 함께 전체를 아우르는 분석이 가능해졌고 새로운 그 무언가를 찾을 기회가 많아졌다. 작년까지는 이런 NGS의 장점과 특성을 살린 연구추세였다면 점차 NGS의 응용범위가 좁은 범위까지 확대되어가는 중이다. Iontorront에 이어 nanopore와 같은 소형 시퀀싱머신의 개발과 보급이 일반화되기 시작했다. 앞으로는 국내의 실험실에도 많은 변화가 있을 것이며, 그 중심은 또 한 번 NGS가 될 것이다. 이젠 국내에서도 bioinformatics의 인식이 달라져야 한다.
작성자 :
(주)인실리코젠 Codes사업본부 Research실유승일 컨설턴트
Impression
- Introduction
- Clustal omega
- Uniprot
- EVA
- GBS
- Single cell analysis















 Figure 2. Exhibitors of ISMB/ECCB2015
Figure 2. Exhibitors of ISMB/ECCB2015

