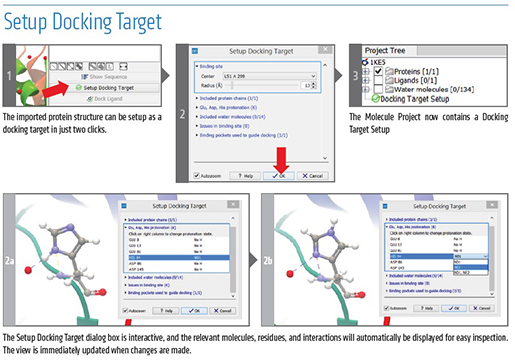
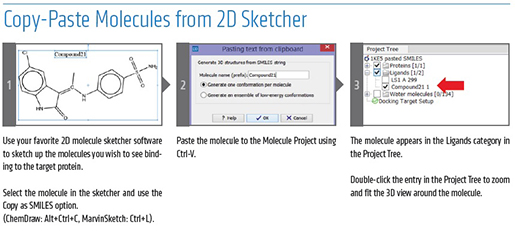
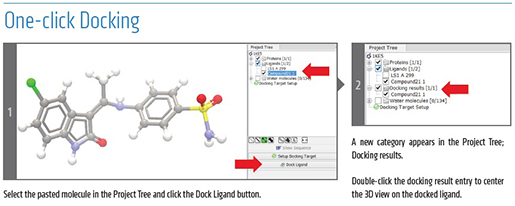
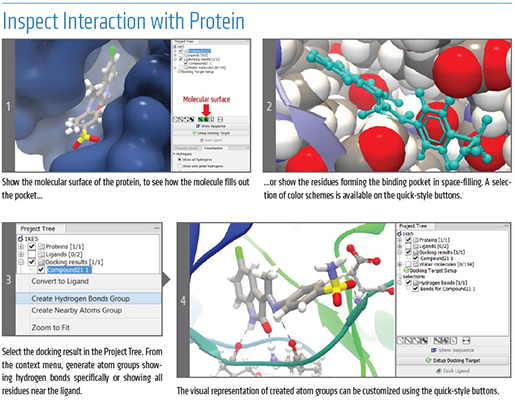
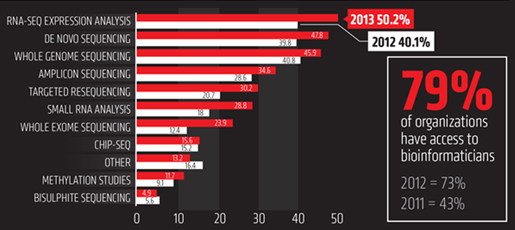
A Superior Solution for Microbial Genomics - 5
- Posted at 2016/08/30 09:55
- Filed under 제품소식

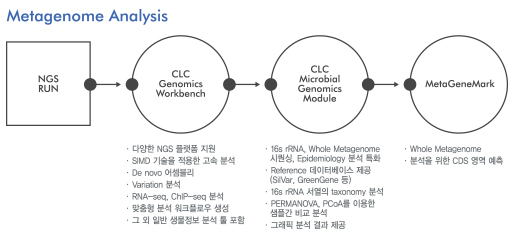
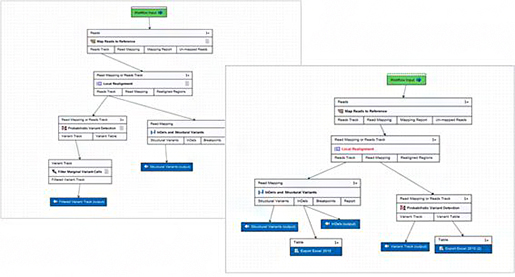
일반 미생물에서부터 난배양성 미생물들까지 모두 확인할 수 있는 방법으로 샘플을 자연상태에서 직접 채취하여 시퀀싱 하는 방법을 metagenome이라고 합니다. NGS가 발전하면서 간단하게 샘플의 16s rRNA를 추출 후 시퀀싱을 하여 해당 샘플내에 존재하는 미생물의 종류와 존재 비율을 알 수 있습니다. CLC Microbial Genomics Module은 16s rRNA 데이터베이스를 다운로드 하는 것부터 OTU clustering, alpha/beta diversity, PERMANOVA 분석까지 가능하게 해주며 미리 구성되어져 있는 워크플로우를 이용해 시퀀싱 raw data를 넣어주는 것만으로 분석이 완료가 됩니다.
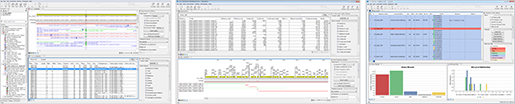
지금부터 보여드릴 데이터는 용의자의 신발 두 켤레에서 나온 흙과 범행현장이라고 예측되는 곳의 토양 샘플의 16s rRNA를 시퀀싱하여 metagenome 분석을 응용한 것입니다. 시퀀싱 데이터를 모듈에 내장되어 있는 'Data QC and OTU Clustering'이라는 워크플로우에 넣어주면 trimming부터 OTU clustering까지 자동으로 진행되게 됩니다.
OTU clustering을 위한 워크플로우
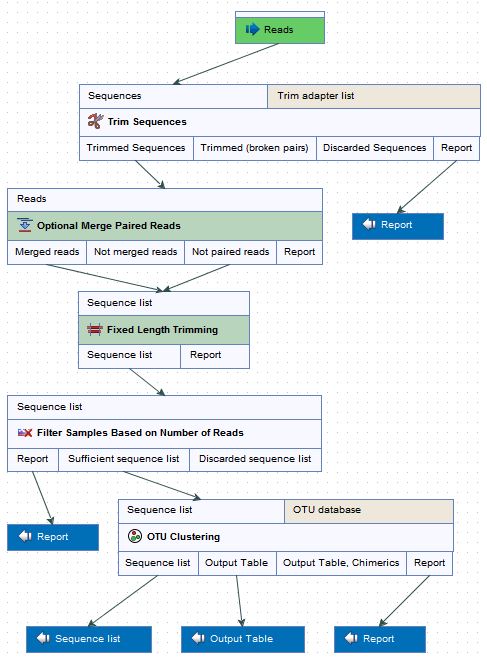
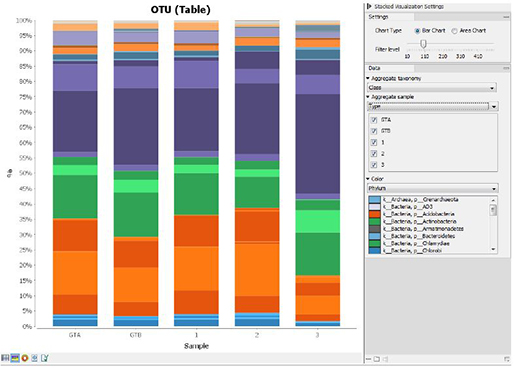
이 워크플로우의 분석 결과로 data trimming report와 OTU clustering 결과를 볼 수 있는데 이 결과는 sunburst chart나 bar chart로 제공됩니다. 각 샘플별 clustering 결과에 metadata를 추가하여 특정 그룹으로 묶어 그룹간의 비교가 가능합니다.
그룹간의 OTU clustering bar chart
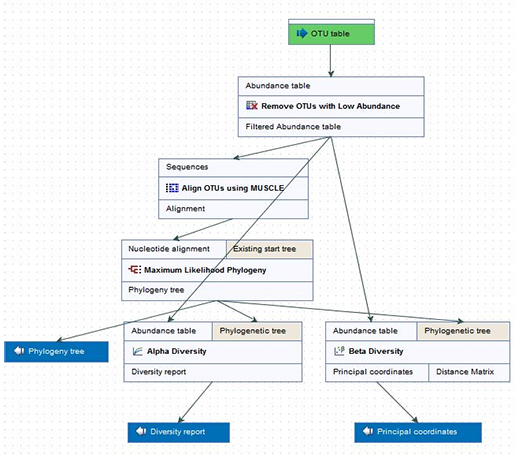
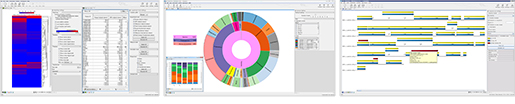
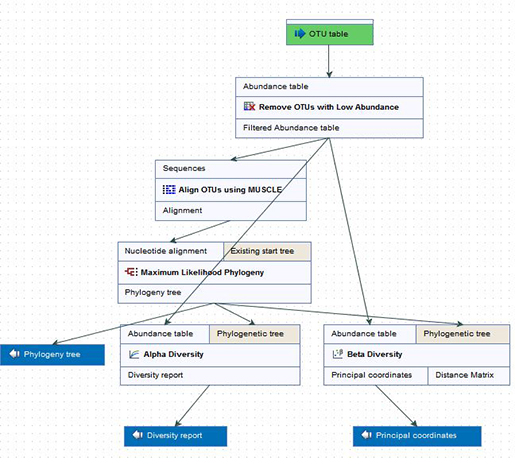
이후 OTU clustering 결과를 가지고 데이터의 taxonomy가 충분히 맵핑 되었는지 확인하기 위해 alpha diversity 분석을 수행하고, 샘플간 혹은 그룹간의 유사도를 보기 위해 beta diversity를 수행하게 됩니다. 그리고 MUSCLE 알고리즘을 이용한 alignment를 진행하고 phylogeny tree를 그려서 각 시퀀스간의 연관성을 확인합니다.
Diversity 확인 및 phylogeny tree 분석을 위한 워크플로우
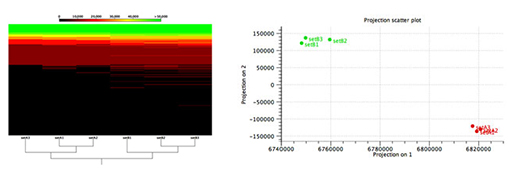
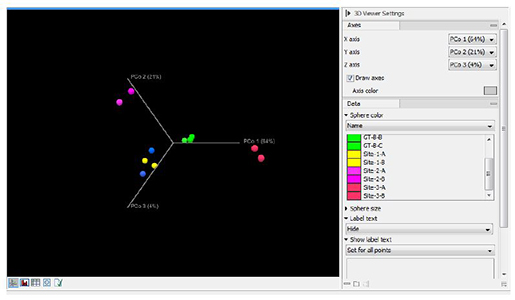
두번째 워크플로우 분석 결과중 하나인 beta diversity의 결과를 함께 봅시다. Metadata를 이용하여 그룹을 지어주면 같은 그룹끼리 같은 색상으로 바뀌게 되며 그룹간 샘플간의 비교분석이 가능합니다. 아래의 그림에서 파란색 동그라미와 노란색 동그라미는 각각 다른 그룹을 의미하지만 유사도의 거리를 따졌을 경우 비슷한 것을 확인 할 수 있습니다 (일치라도 해도 될 정도로 유사함). 따라서 파란색과 노란색은 같은 토양 샘플이라고 잠재적 결정을 내릴 수 있으며, 용의자는 A 부츠를 신고 1번 site에 간 적이 있었다고 결론을 지을 수가 있겠습니다.
Beta diversity 분석 결과
16s rRNA를 이용한 metagenome 분석은 이러한 범인을 찾는데에 응용하는 것 외에도 특정 질병이 잘 걸리는 장내 환경을 조사해볼 수도 있고, 특정 작물이 잘 자라거나 특별히 잘 자라지 않는 토양에서의 미생물 분포를 알아볼 때도 사용할 수 있습니다.
CLC Microbial Genomics Module을 이용하여 우리눈에 보이지 않는 미생물들의 구성과 그 microbial community의 역할 및 특징들을 알아보고 싶으시면 지금 바로 trial 해보세요!
< 이전화 보기 >
(문의) Consulting팀 (대표전화 : 031-278-0061, 이메일 : consulting@insilicogen.com)
Posted by 人Co
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/215

 GFM_tech_note.pdf
GFM_tech_note.pdf