신종 코로나바이러스와 진단키트
- Posted at 2020/04/26 16:36
- Filed under 정보공유

지난 COVID-19에 관한 몇 가지 포스팅에 이어서 오늘은 신종 코로나바이러스(COVID-19) 진단키트에 대하여 알아볼까 합니다.
최근 한국의 바이오 기업이 제조, 생산하는 진단키트들의 높은 정확도와 세계적 찬사를 받으며 그 존재감이 날로 커지고 있는데요, 이탈리아, 스페인, 프랑스, 독일과 같은 유럽 국가를 비롯하여 미국에까지 수출하고 있습니다. 특히 미국 수출건의 경우 까다롭기로 유명한 FDA까지 긴급사용승인을 받아 한국의 진단키트에 대한 대내외적 인지도가 어느 정도인지 가늠해볼 수 있습니다. 이렇듯, 진단키트가 무엇이기에 이토록 한국의 진단키트들이 찬사를 받는지, 신종 코로나바이러스에 진단키트가 중요한 이유가 무엇인지 해당 포스트를 통해 유익한 정보 많이 얻어가시길 바랍니다!

최근 신종 코로나바이러스(SARS-CoV-2)로 인해 많은 사람들이 진단키트에 대한 관심이 높아졌습니다. 진단키트는 각종 질병, 임신 여부, 건강 상태, 친자 확인 등 여러 분야에서 각 목적에 맞게 필요한 시약 및 도구 등을 포함한 생화학 실험 도구를 뜻합니다. 신종 코로나바이러스(SARS-CoV-2)가 전 세계 보건의료를 극심하게 뒤흔들고 있는 상황에서 한국은 진단키트를 통해서 신속 정확하게 진단하여 대처하고 있습니다.
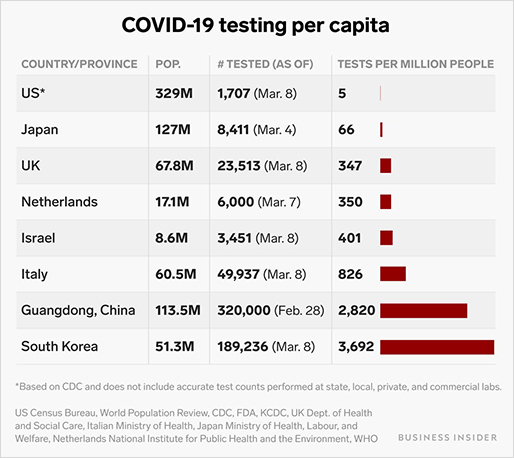
[출처] 신종 코로나바이러스 인구대비 검사율
특히 '방역 한류'라 불릴 정도로 국내에 여러 업체들(씨젠, 솔젠트, 시선바이오 등)에서 제작한 진단키트를 전 세계 곳곳에서 구하려고 힘쓰는 상황입니다. 전 세계에서도 여러 업체가 진단키트를 생산하나 각국의 법적 규제, 생산 라인, 기술 등의 문제로 국내산 진단키트만큼 생산량도 많지 않고 정확도도 낮은 경우가 많습니다. 진단키트에 활용되는 여러 가지 기술은 배양법, 항원-항체 반응, qRT-PCR 등이 있으며 현재 승인이 된 방법은 항원-항체 반응, qRT-PCR입니다.
[출처] qPCR기반의 시선바이오 진단키트
[출처] qPCR기반의 바이오코아 진단키트

[출처] qPCR기반의 씨젠 진단키트

일반적으로, 코로나바이러스는 전염성은 강하지만 병원성이나 치사율은 낮은 바이러스입니다. 그러나 신종 코로나바이러스(SARS-CoV-2)는 전염성도 강하면서 제법 치명적인 병원성을 가지는 특이 변종입니다. SARS-CoV-2의 WHO 공식 치사율은 4월 현재 약 6.7%입니다.
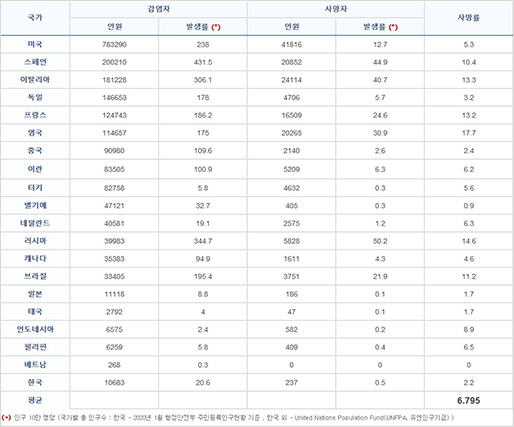
코로나바이러스 중에서 치사율이 1%를 넘기는 사례는 SARS(치사율 9.6%), MERS(치사율 38%) 정도를 제외하면 거의 없습니다. MERS와 SAS가 치사율이 더 높아 보이지만 SARS-CoV-2는 현재 진행형이며, 언제까지 진행될지 예상할 수 없다는 문제가 있습니다. 신종 코로나바이러스에 대한 백신 및 치료제가 개발되지 않은 상황에선 감염자를 빠르고 정확하게 판별해서 격리하고 치료하는 게 최선의 방법입니다.

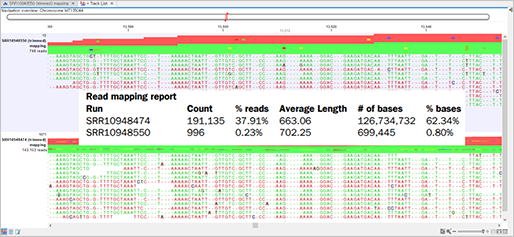
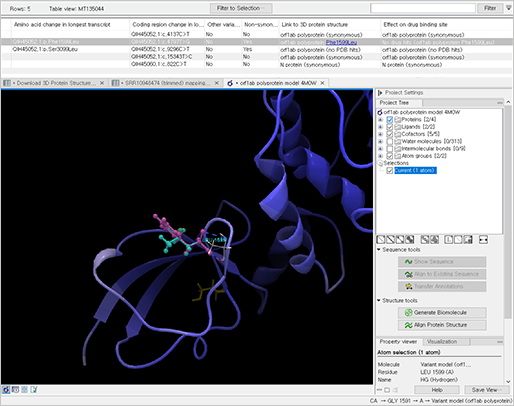
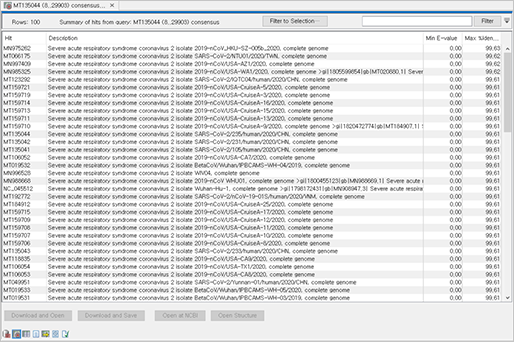
진단키트를 제작하기 위해선 목표하는 질병을 일으키는 세균이나 바이러스 등에서 타 병원체와는 다른 유전자 변이를 찾아내야 합니다. 이를 위해선 생물정보 기술을 활용하여 유전자 변이를 더 쉽게 찾아낼 수 있습니다.

인체 내에 바이러스가 들어올 경우 IgM·G(Immunoglobulin M·G) 항체가 형성됩니다. 신종 코로나바이러스(SARS-CoV-2)가 몸속에 소량이라도 들어오면 이를 방어하기 위해 IgM·G 항체가 생성됩니다. 이렇게 생성된 항체와 결합하는 항원을 통해서 진단할 수 있습니다. 물론 신종 코로나바이러스(SARS-CoV-2)만 특이적으로 검사할 수는 없으나 의심 환자에 대한 광범위한 검사를 진행하여 1차 선별이 가능합니다. 이후 정밀 검사(qRT-PCR등)를 통해 최종 감염 여부를 확인합니다. 1차 선별을 유전자 방식으로만 진행하게 되면 격리된 검사시설과 고가의 장비, 시약, 검사를 수행할 전문 임상 병리사 등이 필요하므로 의심 환자에 대한 광범위한 검사가 어려운 점을 보완할 수 있는 장점이 있습니다.

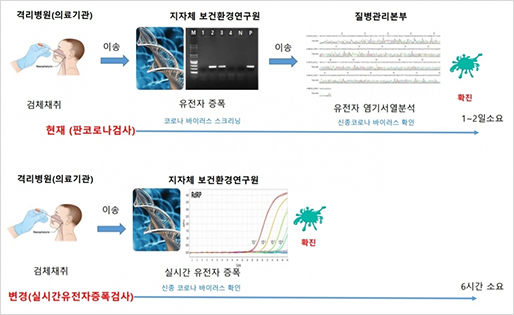
qRT-PCR은 PCR 증폭 산물을 실시간으로 모니터링하는 해석 방법으로, 기존의 PCR 방법으로는 측정하기 어려운 정확한 정량이 가능합니다. 또한, PCR 원리를 기본으로 하고 있으므로 검출감도가 높고, mRNA 발현 해석이나 SNPs typing 등의 유전자 해석에 요구되는 필수 기술입니다. qRT-PCR의 실험 조작은 비교적 간단하며 종래의 PCR법과 거의 유사하다고 생각하면 됩니다. 현재 qRT-PCR을 기반으로 하는 진단키트에 경우 의삼환자에서 객담(가래)을 추출해 코로나19 바이러스가 있는지를 검사하는 방식으로 정확하게 검출할 수 있습니다. 특히 1~2일 걸리는 배양법에 비해 6시간 가량이면 결과를 도출할 수 있어 신속한 진단에 크게 이바지한 방법입니다.
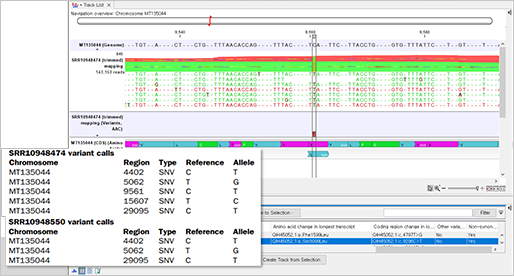
신종 코로나바이러스(SARS-CoV-2)를 특정할 수 있는 유전자 N, E, S, RdRp, Orf1a, Orf1ab 중에서 최소한 두개 이상의 유전자를 증폭하는 Specific primer를 통해서 감염의 여부를 체크할 수 있습니다.
코로나19 환자에게서 위에 나열된 코로나19 바이러스의 모든 유전자를 완전히 다 검출해서 완벽하게 대조하면 좋겠지만, 그건 오래 걸리고 비용면에서도 효율성이 떨어지기 때문입니다. 그래서 국가마다 검출 유전자는 조금씩 다르지만, 보통은 가장 변이를 덜 일으킬 것으로 보이는 유전자를 최소 2개 이상 검사해서 둘 다 '양성' 반응이 나타나는지를 보고 코로나19 감염 여부를 판단하고 있는 겁니다.

신종 코로나바이러스(SARS-CoV-2) 감염증의 사례로 알 수 있듯이 진단키트를 활용하여 빠르게 감염자를 선별하고 격리하여 2차 피해를 예방하고 조기에 치료하는 방법이 제일 좋은 방법입니다. 또한, 병원체 (바이러스, 세균 등)은 빠르게 변이되고 돌연변이를 통해 또 다른 질병을 일으킬 수 있으므로 이에 우리는 생물정보 기술을 통해 신속하게 병원체를 연구 및 분석하여 기존의 병원체와의 차이점을 찾아내고 진단키트로 제작하여 대처해야 합니다.
현재 (주)인실리코젠에서는 신종 코로나바이러스(SARS-CoV-2) 연구를 포함한 감염병 연구를 위해 생물정보 솔루션인 CLC Genomics ProSuite(CLC Genomics Workbench를 포함한 바이러스 및 미생물 NGS 분석 패키지)와 Ingenuity Pathway Analysis(IPA, 유전자 네트워크 분석 솔루션)의 단기 라이선스를 지원하고 있으니 신청하시면 6월 15일까지 사용해보실 수 있습니다.


[출처] 항원항체반응진단원리
qRT-PCR은 PCR 증폭 산물을 실시간으로 모니터링하는 해석 방법으로, 기존의 PCR 방법으로는 측정하기 어려운 정확한 정량이 가능합니다. 또한, PCR 원리를 기본으로 하고 있으므로 검출감도가 높고, mRNA 발현 해석이나 SNPs typing 등의 유전자 해석에 요구되는 필수 기술입니다. qRT-PCR의 실험 조작은 비교적 간단하며 종래의 PCR법과 거의 유사하다고 생각하면 됩니다. 현재 qRT-PCR을 기반으로 하는 진단키트에 경우 의삼환자에서 객담(가래)을 추출해 코로나19 바이러스가 있는지를 검사하는 방식으로 정확하게 검출할 수 있습니다. 특히 1~2일 걸리는 배양법에 비해 6시간 가량이면 결과를 도출할 수 있어 신속한 진단에 크게 이바지한 방법입니다.
[출처] 배양법-qPCR기반진단법 차이
신종 코로나바이러스(SARS-CoV-2)를 특정할 수 있는 유전자 N, E, S, RdRp, Orf1a, Orf1ab 중에서 최소한 두개 이상의 유전자를 증폭하는 Specific primer를 통해서 감염의 여부를 체크할 수 있습니다.
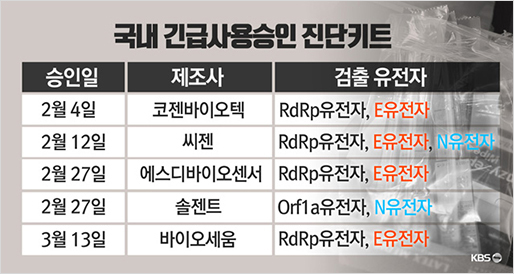
[출처] 국내 긴급사용승인 진단키트
코로나19 환자에게서 위에 나열된 코로나19 바이러스의 모든 유전자를 완전히 다 검출해서 완벽하게 대조하면 좋겠지만, 그건 오래 걸리고 비용면에서도 효율성이 떨어지기 때문입니다. 그래서 국가마다 검출 유전자는 조금씩 다르지만, 보통은 가장 변이를 덜 일으킬 것으로 보이는 유전자를 최소 2개 이상 검사해서 둘 다 '양성' 반응이 나타나는지를 보고 코로나19 감염 여부를 판단하고 있는 겁니다.
신종 코로나바이러스(SARS-CoV-2) 감염증의 사례로 알 수 있듯이 진단키트를 활용하여 빠르게 감염자를 선별하고 격리하여 2차 피해를 예방하고 조기에 치료하는 방법이 제일 좋은 방법입니다. 또한, 병원체 (바이러스, 세균 등)은 빠르게 변이되고 돌연변이를 통해 또 다른 질병을 일으킬 수 있으므로 이에 우리는 생물정보 기술을 통해 신속하게 병원체를 연구 및 분석하여 기존의 병원체와의 차이점을 찾아내고 진단키트로 제작하여 대처해야 합니다.
현재 (주)인실리코젠에서는 신종 코로나바이러스(SARS-CoV-2) 연구를 포함한 감염병 연구를 위해 생물정보 솔루션인 CLC Genomics ProSuite(CLC Genomics Workbench를 포함한 바이러스 및 미생물 NGS 분석 패키지)와 Ingenuity Pathway Analysis(IPA, 유전자 네트워크 분석 솔루션)의 단기 라이선스를 지원하고 있으니 신청하시면 6월 15일까지 사용해보실 수 있습니다.
하루 속히 신종 코로나바이러스(SARS-CoV-2) 감염 확산이 종식되길 바라며, 앞으로 이러한 생물정보 솔루션들의 활용을 통해서 바이러스 및 병원체 발생을 조기에 감지하고 제어하는 공중 보건의 보호에 도움이 되길 바랍니다.
Posted by 人Co
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/343