
Assembly를 해보자!
- Posted at 2012/04/13 09:36
- Filed under 제품소식
"저는 생물학과를 졸업 후 분자생물학 실험을 주로 하는 실험실에 석사과정으로 들어간 대학원생입니다. 여기서 저의 막내 생활이 다시 시작됩니다.
어느 날 교수님께서 '너의 석사 주제를 가져왔노라'시며 의미심장한 미소를 띄우시고는 미팅룸으로 저를 끌고 가십니다.
요즘 NGS 라는게 뜨는데 네가 남자고 컴퓨터를 좀 더 잘 할 터이니 네가 이걸로 뭔가를 해보라고 하십니다.
그 '뭔가'가 대체 뭔지는 논문 찾아보면 다 나오니까 조사해오라고 하십니다.
나름 조사를 해보니 용어도 잘 모르겠고, 다 영어라 해석도 어렵고...
전 정말 미추어 버리겠지만, 어쩌겠습니까? 까라면 까야죠."
위의 이야기는 제가 아는 어떤 친구의 하소연입니다. 이런 상황은 이 친구 뿐만이 아니라 대한민국 실험실의 많은 학생들, 그리고 연구원들이 공감하는 이야기일 것 같습니다.
저도 그 답답함을 겪었던, 그리고 아직 겪고 있는 한 사람으로서 제가 아는 만큼 NGS에 대해서 쉽게 이해할 수 있는, 그리고 소통을 통해 그 답답함을 해결할 수 있게 하는 글을 남겨보고자 합니다. 자 그럼 NGS 정체에 대해서부터 이야기 해보겠습니다.
NGS가 뭡니까?
NGS는 Next Generation Sequencing의 약자로 차세대 염기서열 결정이라고 해석할 수 있겠습니다. 해석은 ‘염기서열 결정’이라고 했는데 ‘휴먼 지놈 시퀀싱’ 할 때의 그 시퀀싱을 쓰는게 더 어울릴 것 같네요. 아무튼 어떤 생명체의 염기 서열을 알아낼 때 쓰는 시퀀싱법의 최신 버젼을 말하는 것 같습니다. 차세대라고 하는걸 보면 이전 세대의 염기서열 결정법 보다 뭔가 더 좋은게 있는 것 같습니다. 얼마나 혁신적으로 좋아졌길래 ‘차세대’라는 말을 갖다 붙였을까요?
'이전 세대' 시퀀싱법 Sanger method
Sanger method는 학부 분자생물학 시간 때 언뜻 들은 기억이 납니다. 지놈을 무작위로 잘라 단편 조각으로 만들고, single strand로 만든 다음 PCR처럼 primer를 붙이고 중합효소로 상보적인 dNTP들을 붙여 나갈 때 형광다이가 붙어 있는 ddNTP가 붙으면 중합 반응이 끝나고, 그 조각들을 전기영동으로 분리하면 짧은 조각부터 1bp 간격으로 정렬이 되는데 이 순서대로 형광다이의 색으로 A, T, G, C 서열을 결정해 나가는 방법입니다. 비루한 저의 설명 보다는 없는 지식이 없는 위키피디아(http://en.wikipedia.org/wiki/DNA_sequencing)나 파워블로거 님들의 자료를 살펴 보심이 옳은 줄로 아뢰옵니다. Sanger method의 장점이라면 정확해서 믿을만하다고 할 수 있겠습니다. 단점은 비싸고 오래 걸린다는 점입니다. 그래서 연구용으로는 적합하지만 상업적 목적으로 이용하기에는 힘들다고 합니다.
그럼 NGS는 뭐가 좋은데?
싸고 빠릅니다. 어디까지나 Sanger method에 비해서라는 전제가 붙을 때 이야기입니다. 차세대 염기서열 결정법은 크게 4~5가지가 있습니다. 기존 Sanger method의 비싸고 오래 걸려서 상업적으로 이용하기 어렵다는 단점을 극복할 수 있는 시퀀싱법을 개발하기 위해 몇몇 회사가 뛰어들어 새로운 시퀀싱법을 개발해 냈는데 기본적으로 전체 genome을 잘라 단편으로 만든 후 각 단편의 서열을 시퀀싱하고 이 단편들을 assembly라는 단계에서 겹쳐지는 부분을 이어 원래 지놈 서열을 알아내는 식으로 genome project가 진행 됩니다. 각 회사마다 개발해낸 시퀀싱법이 적용된 장비(혹은 플랫폼이라고 부르기도 합니다.)를 개발해서 판매하고 있으며 대표적으로 Roche사의 GS-FLX, Illumina의 Genome Analyzer, Applied Biosystem사의 SOLiD, Life Technologies사의 Ion Torrent 등이 있습니다. 여기서부터 슬슬 복잡해지기 시작합니다. 각 장비들마다 다른 시퀀싱법을 사용하다 보니 저마다 각자의 강점과 약점이 있습니다.
NGS로 무엇을 할 수 있을까?
서열을 시퀀싱해서 할 수 있는 분석들 대부분을 할 수 있습니다. 크게 나누자면 genomics, transcriptomics, epigenomics 분야로 나눌 수 있지만 raw data를 어떻게 응용하느냐에 따라 더 다양한 분야의 분석 방법들이 만들어 질 수 있을 것 같습니다.
NGS 분석을 위해 준비해야 할 것들
"자 그럼 NGS 분석을 해보자! 뭐부터? 설계 먼저!
헐~ 데이터 파일이 너무 큰데... 내 PC에서 하면 PC 폭발하는거 아녀?
받긴 받았는데.. 이게 다 뭐다냐?"
어느 실험이 다 그렇듯이 NGS 데이터를 분석하는 것도 정확한 계획을 세우는 것이 중요합니다. 그 계획을 세우기 위해서는 시퀀싱 대상이 원핵 생물인지 진핵 생물인지? 지놈의 크기는 어느 정도인지? 그리고 얼마나 많이 시퀀싱 할 것인지(depth)? 그리고 주로 분석 할 대상이 RNA서열인지? exom 서열인지? whole 지놈 서열인지? 와 같이 시퀀싱 대상에 대한 정보를 파악하고 분석 목적에 맞는 시퀀싱 데이터(single short, long read, paired-end read, mate paired-end read)를 만드는 것이 중요합니다. 그리고 이 데이터들을 분석하기 위한 고사양의 컴퓨터, 분석 소프트웨어도 준비해야 합니다. 이 부분에 대한 설명은 다음으로 넘기도록 하겠습니다.
"de novo" assembly를 해보자!
'de novo' 라는 말이 낯섭니다. 우리 친구 네이놈 사전을 찾아보니 부사로써 ’처음부터‘, ’새로(이)(anew)‘, ’다시(again)‘, 영영사전에는 'from the beginning' 이라는 뜻이랍니다. 그러니까 de novo assembly는 새롭게 assembly를 한다는 말이 되겠네요. de novo assembly는 주로 서열이 밝혀지지 않은 종의 지놈 서열을 알아내고자 할 때 수행하는 분석입니다. Assembly는 일종의 퍼즐 맞추기랑 비슷합니다. Reference assembly는 원본 사진을 보면서 맞추는 퍼즐이고 de novo assembly는 원본 사진 없이 맞추는 거라고 보시면 됩니다.
 * 1,000 피스 퍼즐 맞추는데 3일이 넘게 걸린다는데 이 퍼즐은 18,235 피스랍니다. 단순 비례 계산으로 치면 54일 쯤 걸렸겠네요.
* 1,000 피스 퍼즐 맞추는데 3일이 넘게 걸린다는데 이 퍼즐은 18,235 피스랍니다. 단순 비례 계산으로 치면 54일 쯤 걸렸겠네요.
원본 없이 하나하나 맞춰보면서 연결되는 것들 끼리 쭉~ 이어 원래의 genome 서열을 알아내야 하는 작업인지라 한 종류의 read들만 가지고 assemble을 하기란 쉽지 않습니다. 그래서 short, long, paired-end, mate-paired-end 등 여러 종류의 read들을 가지고 assembly를 해야 결과가 좋아집니다.
 * 과학시간에 많이 보던 큰 구슬, 작은 구슬로 비커 채우기 실험
* 과학시간에 많이 보던 큰 구슬, 작은 구슬로 비커 채우기 실험
NGS read 데이터를 다운받자!
Assembly를 하려면 먼저 NGS read가 필요합니다. 현재 NCBI, EBI, DDBJ의 sequence 데이터 저장 서비스인 SRA(Sequence Read Archive)에서 공개된 NGS 데이터를 받을 수 있습니다. 일단 이 블로그에서는 어떻게 de novo assembly를 돌릴 수 있는지 알아보는 것이 목적이므로 지놈의 크기가 비교적 작은 E. coli의 NGS 데이터를 사용하고자 합니다. 웹 브라우저에서 아래 url로 이동해 보시면 CLC bio에서 제공하는 예제 데이터로 E.coli의 genome을 sequencing한, Roche의 454 장비에서 만들어진 long read 데이터와, Illumina 장비에서 만들어진 paired-end read 데이터를 다운받을 수 있습니다.
http://www.clcbio.com/index.php?id=1290

Roche 454 데이터 불러오기
Roche의 데이터는 크게 두 가지 형태가 있습니다. 하나는 454 Flowgram 이라고 불리는 .sff 파일이고 다른 하나는 FASTA 포맷으로 서열과 그에 대응하는 quality 값이 각각 두 개의 파일에 분리된 형태가 있습니다. 여기서 사용할 데이터는 후자에 해당합니다. 먼저 다운로드 받은 Roche 데이터의 압축 파일을 풀면 3개의 파일이 생성됩니다. 하나는 이미 알려진 E.coli의 전체 genome 서열 파일(.gbk)이고 다른 두 파일에 NGS read 파일입니다. 이중에서 .fna 파일이 각 read의 서열이 있는 파일이고 .qual 파일은 각 서열의 quality 값이 순서대로 기록된 파일입니다. 그 리고 CLC Genomics Workbench 화면 위쪽의 Tool bar에서 'NGS import'라는 버튼을 클릭하면 다음과 같이 import 할 수 있는 NGS data의 종류가 나타납니다. 여기서 첫 번째에 있는 'Roche 454...‘를 클릭합니다.
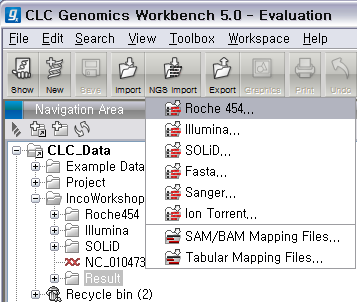 검색위치에서 아까 압축을 풀었던 폴더를 찾아간 후 .fna 파일과 .qual 파일을 둘 다 선택한 후 'Next'를 클릭하고,
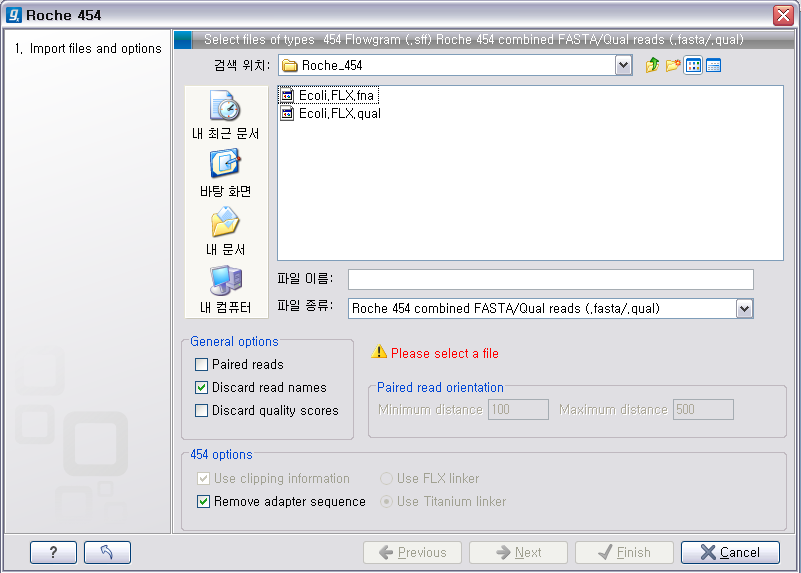
다음 화면에서 저장위치 선택 후 'Finish' 버튼을 클릭하면 import가 완료 됩니다. Roche의 장비에서 나오는 서열에는
adapter sequence라는게 존재합니다. 이 adapter 서열은 원래 시퀀싱을 한 sample에서 나온 서열이 아니기
때문에 제거해 주는 과정이 필요한데 '454 options'에 있는 'Remove adapter sequence' 옵션이 항상
체크되어 있어야 합니다.
검색위치에서 아까 압축을 풀었던 폴더를 찾아간 후 .fna 파일과 .qual 파일을 둘 다 선택한 후 'Next'를 클릭하고,
다음 화면에서 저장위치 선택 후 'Finish' 버튼을 클릭하면 import가 완료 됩니다. Roche의 장비에서 나오는 서열에는
adapter sequence라는게 존재합니다. 이 adapter 서열은 원래 시퀀싱을 한 sample에서 나온 서열이 아니기
때문에 제거해 주는 과정이 필요한데 '454 options'에 있는 'Remove adapter sequence' 옵션이 항상
체크되어 있어야 합니다.
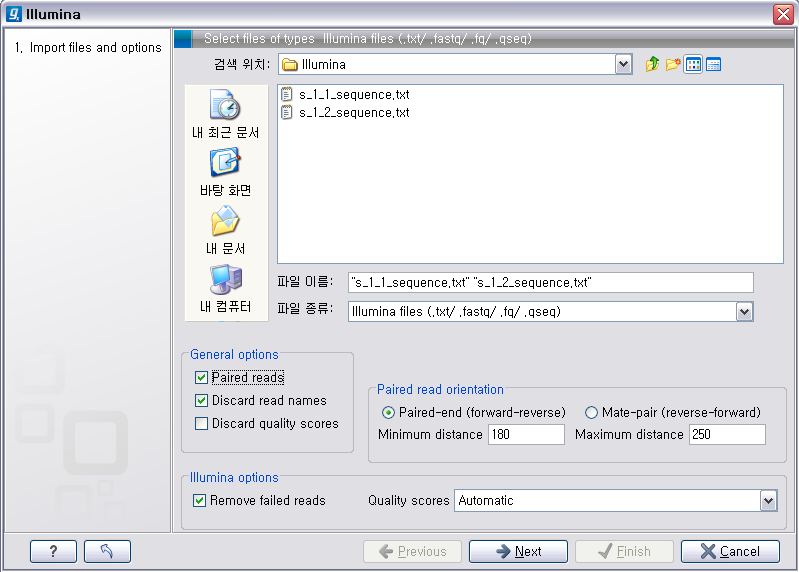
Illumina 데이터 불러오기
이번에 불러올 Illumina의 데이터는 paired-end read입니다. 검색위치에서 Illumina 데이터의 압축을 풀어놓은 폴더를 찾아간 후 forward 방향의 read 파일과 reverse 방향의 read파일을 둘다 선택 하시고, ‘General options'에 있는 ’Paired reads'라는 옵션을 체크해 줘서 이 read들이 paired-end read임을 인식하도록 해줍니다. 'Paired read' 옵션이 선택되면 ‘Paired read orientation'이라는 항목에 대한 옵션을 조정 할 수 있도록 활성화가 되는데 여기서 insertion size를 조정해 줍니다. 그리고 ’Next' 버튼을 클릭하고 저장할 위치를 설정한 후 ‘Finish' 버튼을 클릭하면 import가 완료됩니다.
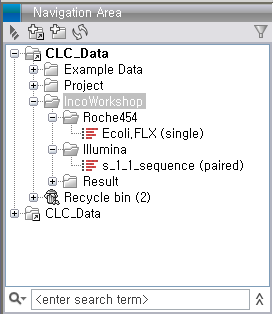
de novo assembly 돌리기
Import가 완료되면 다음 그림과 같이 Roche와 Illumina 데이터가 각각 하나씩 나타나게 됩니다. Illumina 데이터의 파일은 두 개였지만 import 가 되면서 하나의 데이터로 합쳐진 것을 볼 수 있습니다.
 데이터 준비가 완료되면 'Toolbox'에서 'High-Throughput Sequencing'에 있는 'De Novo
Assembly'라는 툴을 더블클릭하여 실행 시킵니다. 첫 번째 화면에서 assemble 할 read 데이터를 선택하고
'Next'를 클릭합니다.
데이터 준비가 완료되면 'Toolbox'에서 'High-Throughput Sequencing'에 있는 'De Novo
Assembly'라는 툴을 더블클릭하여 실행 시킵니다. 첫 번째 화면에서 assemble 할 read 데이터를 선택하고
'Next'를 클릭합니다.
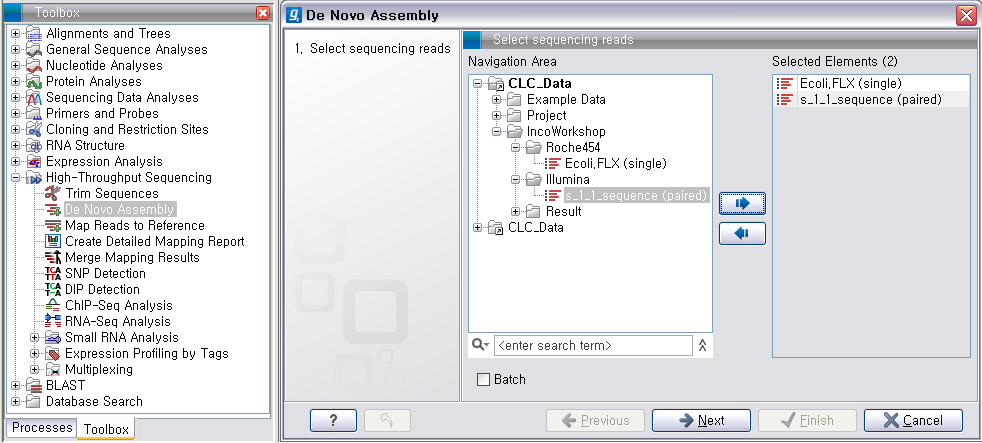 그 다음 단계에서는 de novo assembly를 하는데 필요한 몇 가지 옵션 값들을 선택하도록 되어 있습니다. CLC
Genomics Workbench는 de Bruijn graph 라는 알고리즘으로 assembly를 합니다. 이 때 원래
read들을 더 작은 단편들로 만드는데, 이 단편들의 길이를 word size라고 부릅니다. ‘Automatic word
size’를 체크하면 데이터의 크기에 따라서 정해진 word size로 assembly를 하게 됩니다. ‘Guidance only reads’는 scaffolding에 사용할 read를 설정하는 옵션으로 mate paired-end read를 선택해야 합니다. ‘Contig
length’는 assembly된 contig 서열의 최소 길이를 설정하는 옵션이고, 'Perform scaffolding'은
만들어진 contig들과 가이드로 사용할 paired-end read를 가지고 더 큰 contig인 scaffold 서열을 만들
것인지 설정하는 옵션입니다. de novo assembly 옵션들의 설정이 완료되면 ‘Next'를 클릭합니다.
그 다음 단계에서는 de novo assembly를 하는데 필요한 몇 가지 옵션 값들을 선택하도록 되어 있습니다. CLC
Genomics Workbench는 de Bruijn graph 라는 알고리즘으로 assembly를 합니다. 이 때 원래
read들을 더 작은 단편들로 만드는데, 이 단편들의 길이를 word size라고 부릅니다. ‘Automatic word
size’를 체크하면 데이터의 크기에 따라서 정해진 word size로 assembly를 하게 됩니다. ‘Guidance only reads’는 scaffolding에 사용할 read를 설정하는 옵션으로 mate paired-end read를 선택해야 합니다. ‘Contig
length’는 assembly된 contig 서열의 최소 길이를 설정하는 옵션이고, 'Perform scaffolding'은
만들어진 contig들과 가이드로 사용할 paired-end read를 가지고 더 큰 contig인 scaffold 서열을 만들
것인지 설정하는 옵션입니다. de novo assembly 옵션들의 설정이 완료되면 ‘Next'를 클릭합니다.
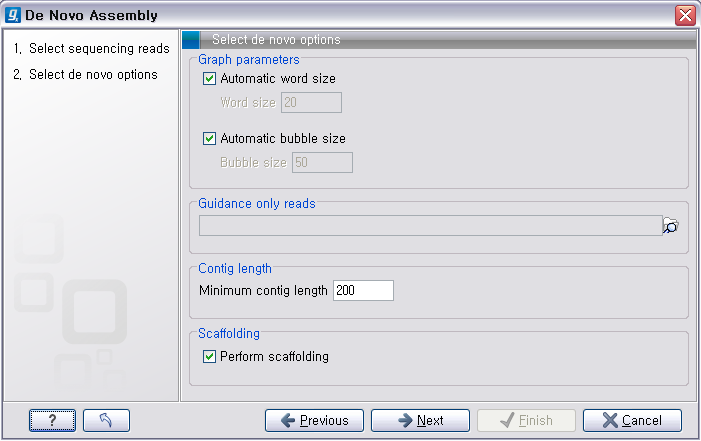 다음 단계의 옵션들은 mapping에 관련된 옵션들입니다. de novo assembly 하고 나면 일단 contig 서열들만
만들어 지게 됩니다. 이 contig 서열을 reference로 하여 read들을 mapping 시켜서 각 contig가 어떤
read들의 조합으로 만들어진 것인지 알 수 있게 합니다. ‘Update contigs' 옵션을 체크하게 되면 contig에 read가 mapping되는 정보가 contig 서열에 반영되게 됩니다. ‘Create simple contig sequences'를 선택하고 ’Next'를 클릭하면 이러한 과정 없이 contig 서열 들만 만들어 내게 됩니다. 옵션 설정이 완료되면 'Next'를 클릭하고 저장위치를 설정한 후 'Finish' 버튼을 클리하면 de novo assembly가 시작됩니다.
다음 단계의 옵션들은 mapping에 관련된 옵션들입니다. de novo assembly 하고 나면 일단 contig 서열들만
만들어 지게 됩니다. 이 contig 서열을 reference로 하여 read들을 mapping 시켜서 각 contig가 어떤
read들의 조합으로 만들어진 것인지 알 수 있게 합니다. ‘Update contigs' 옵션을 체크하게 되면 contig에 read가 mapping되는 정보가 contig 서열에 반영되게 됩니다. ‘Create simple contig sequences'를 선택하고 ’Next'를 클릭하면 이러한 과정 없이 contig 서열 들만 만들어 내게 됩니다. 옵션 설정이 완료되면 'Next'를 클릭하고 저장위치를 설정한 후 'Finish' 버튼을 클리하면 de novo assembly가 시작됩니다.
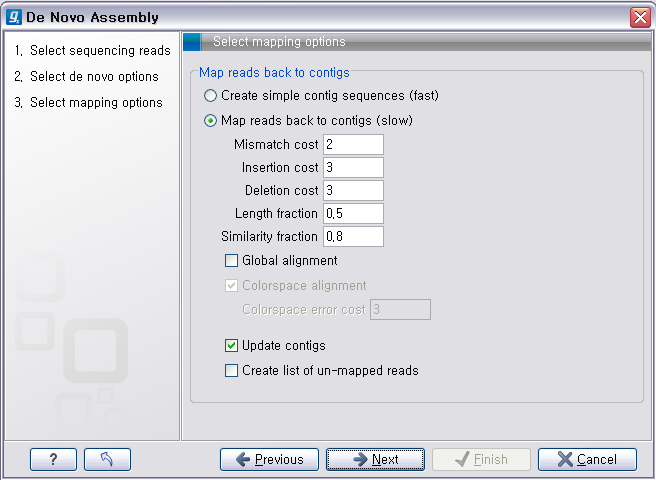 de novo assembly의 결과 데이터를 열어보면 다음과 같은 테이블을 볼 수 있습니다. 이 테이블은 각 de novo
assembly의 결과로 만들어진 각 contig들의 정보를 보여 주며, 생성된 contig의 수, 각 contig의 길이, 각
contig에 mapping된 read의 수, 각 contig의 평균 coverage와 같은 정보를 알 수 있게 됩니다.
de novo assembly의 결과 데이터를 열어보면 다음과 같은 테이블을 볼 수 있습니다. 이 테이블은 각 de novo
assembly의 결과로 만들어진 각 contig들의 정보를 보여 주며, 생성된 contig의 수, 각 contig의 길이, 각
contig에 mapping된 read의 수, 각 contig의 평균 coverage와 같은 정보를 알 수 있게 됩니다.
 각 항목을 더블클릭하면 새 데이터 탭이 열리면서 해당 contig와 그 contig에 mapping된 read들의 mapping
view가 나타나게 됩니다. 가운데 가는 실선으로 이어진 굵은 파란색 선은 paired-end read를 나타내고 연두색 선은
forward 방향의 single read, 빨간색 선은 reverse 방향으로 mapping 된 single read를
의미합니다. 각 read에서 세로 방향으로 그어진 작은 선들은 contig 서열과 다른 base로 conflict라고 부르며 A,
T, G, C 각 염기 마다 다른 색깔로 표시 됩니다. de novo assembly를 할 때 이런 conflict가 많을 경우
sequencing 에러가 높다고 의심해 볼 수 있으며, 혹은 heterozygous 종의 지놈을 시퀀싱 했다고 추정해 볼 수
있습니다.
각 항목을 더블클릭하면 새 데이터 탭이 열리면서 해당 contig와 그 contig에 mapping된 read들의 mapping
view가 나타나게 됩니다. 가운데 가는 실선으로 이어진 굵은 파란색 선은 paired-end read를 나타내고 연두색 선은
forward 방향의 single read, 빨간색 선은 reverse 방향으로 mapping 된 single read를
의미합니다. 각 read에서 세로 방향으로 그어진 작은 선들은 contig 서열과 다른 base로 conflict라고 부르며 A,
T, G, C 각 염기 마다 다른 색깔로 표시 됩니다. de novo assembly를 할 때 이런 conflict가 많을 경우
sequencing 에러가 높다고 의심해 볼 수 있으며, 혹은 heterozygous 종의 지놈을 시퀀싱 했다고 추정해 볼 수
있습니다.
Reference assembly를 해보자!
앞에서 reference assembly가 원본 사진을 보고 퍼즐을 맞추는 것과 비슷하다고 말했습니다. Reference assembly는 말 그대로 reference가 되는 원본 서열에 read들을 mapping시켜 만들어지는 consensus 서열을 얻는 것을 말합니다. 예를 들어 한우의 유전체를 시퀀싱 하여 얻어낸 read 데이터를 NCBI에 공개된 소의 reference 서열에 mapping 시켜서 한우의 유전체와 어떤 차이가 있는지 비교하는 분석을 수행할 수 있을 것입니다.
이번에도 Roche 454 데이터와 Illumina의 데이터를 이용하되, 함께 압축되어 있던 E.coli 지놈 서열(NC_010473)을 reference로 두고 mapping 시켜 보도록 하겠습니다. Reference 서열 파일, NC_010473.gbk는 마우스로 클릭앤드래그하여 ‘Navigation Area’에 옮기면 자동으로 import가 완료 됩니다.
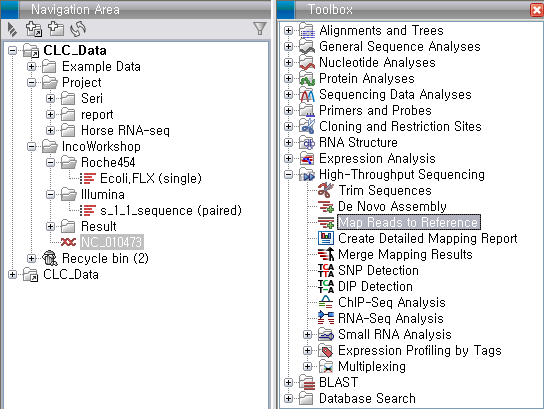 'Toolbox'에서 'High-Throughput Sequencing'에 있는 'Map Reads to Reference'라는
툴을 더블클릭하여 실행 시킵니다. 첫 번째 화면에서 assemble 할 read 데이터를 선택하고 'Next'를 클릭합니다.
'Toolbox'에서 'High-Throughput Sequencing'에 있는 'Map Reads to Reference'라는
툴을 더블클릭하여 실행 시킵니다. 첫 번째 화면에서 assemble 할 read 데이터를 선택하고 'Next'를 클릭합니다.
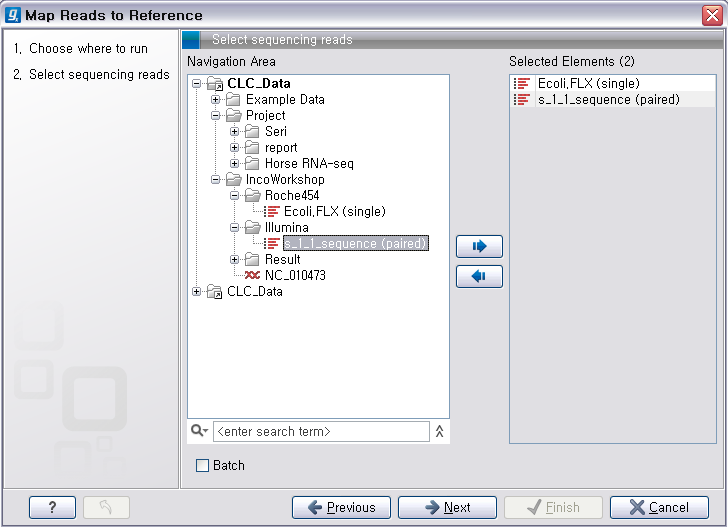 다음 단계에서는 reference가 될 서열을 선택해 줍니다. 이 때 여러 개의 reference 서열을 지정할 수 있습니다.
예를 들어 human의 NGS 데이터를 전체 염색체에 mapping 시키고자 할 경우 22개의 상염색체와 2개의 성염색체, 그리고
필요에 따라 mitochondrial 염색체 서열을 선택해야 합니다. 선택이 완료되면 ‘Next'를 클릭합니다.
다음 단계에서는 reference가 될 서열을 선택해 줍니다. 이 때 여러 개의 reference 서열을 지정할 수 있습니다.
예를 들어 human의 NGS 데이터를 전체 염색체에 mapping 시키고자 할 경우 22개의 상염색체와 2개의 성염색체, 그리고
필요에 따라 mitochondrial 염색체 서열을 선택해야 합니다. 선택이 완료되면 ‘Next'를 클릭합니다.
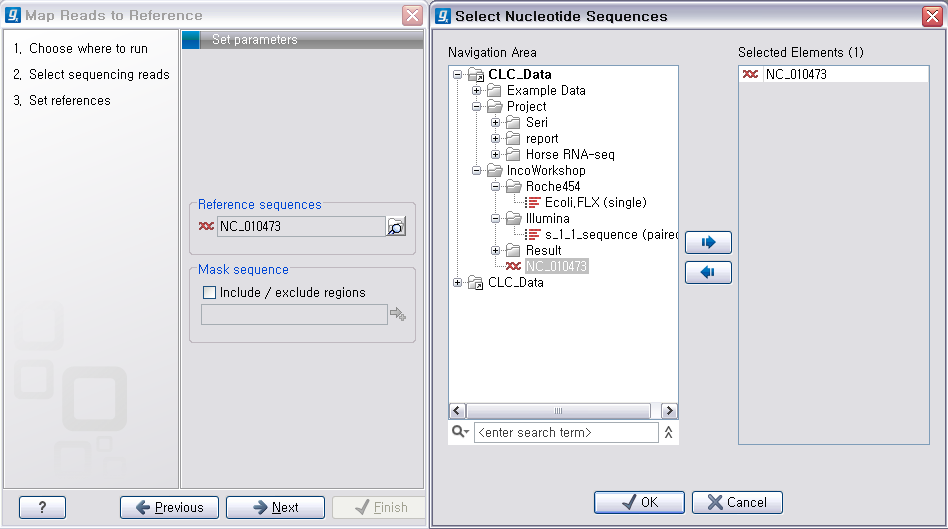 다음 단계에서는 mapping에 관련된 옵션들을 선택합니다. 크게 long read에 대하여 설정해야 하는 옵션과 short read 대하여 설정해야 하는 옵션으로 구분됩니다.
다음 단계에서는 mapping에 관련된 옵션들을 선택합니다. 크게 long read에 대하여 설정해야 하는 옵션과 short read 대하여 설정해야 하는 옵션으로 구분됩니다.
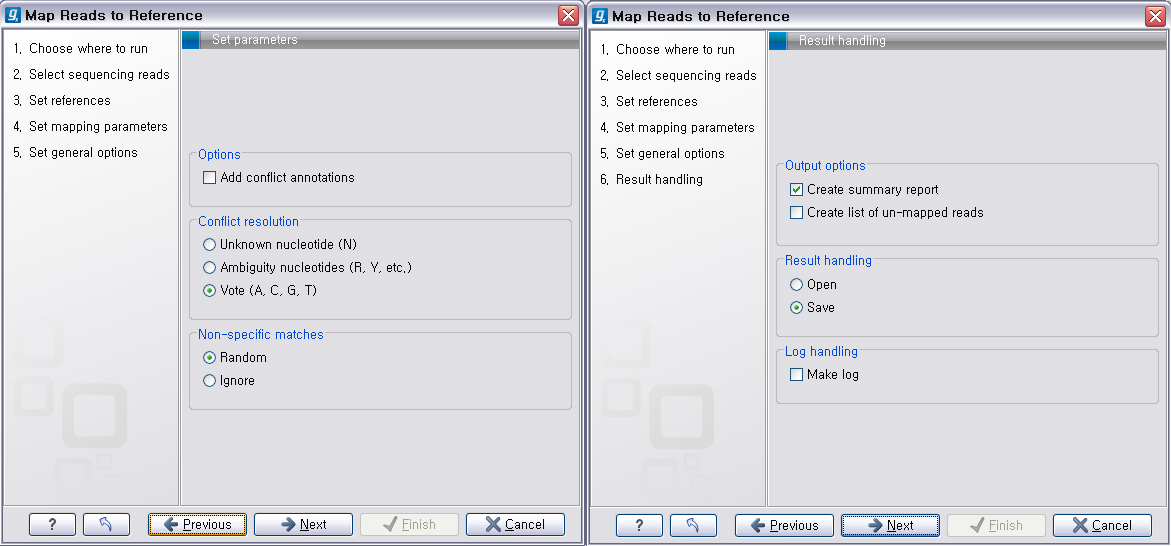
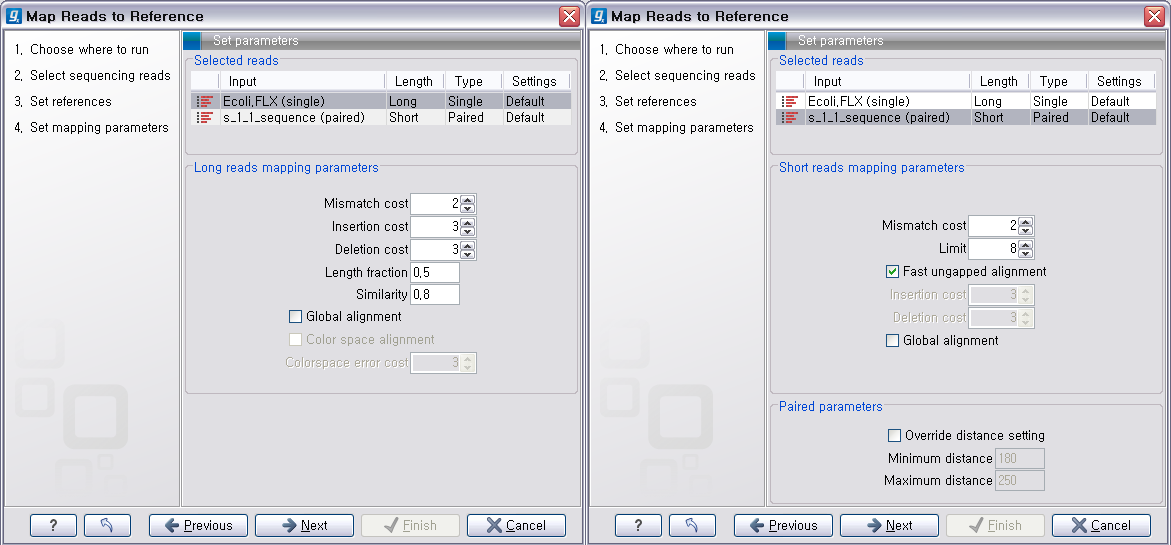 다음 단계에서는 일반적인 결과 처리에 관련된 옵션들을 설정합니다. 'Add conflict annotation'을 클릭하면
consensus서열과 다른 reference 서열의 염기에 'Conflict' 라는 annotation을 입혀 주게 됩니다.
reference 서열의 크기가 크고 sequencing depth가 높을수록 비교적 conflict가 많이 발생하게 됩니다. 이
때 이 옵션의 설정을 체크하면 conflict 부분을 가시화 하는데 상당히 많은 메모리를 필요하게 되어 결과를 보는데 많이
시간이 필요하게 될 수 있습니다. 다음 옵션인 'Conflict resolution'은 conflict가 발생한 부분의 consensus 염기를 어떻게 결정할 것인지를 설정하는 옵션입니다. 그
리고 'Non-specific matches', 즉 reference 서열에 특정 부분에만 붙지 않고 다수의 부분에 붙는
read를 ‘Random'하게 붙일 것인지 ’Ignore'(무시) 할 것인지 설정 한 후 'Next'를 클릭합니다.
다음 단계에서는 일반적인 결과 처리에 관련된 옵션들을 설정합니다. 'Add conflict annotation'을 클릭하면
consensus서열과 다른 reference 서열의 염기에 'Conflict' 라는 annotation을 입혀 주게 됩니다.
reference 서열의 크기가 크고 sequencing depth가 높을수록 비교적 conflict가 많이 발생하게 됩니다. 이
때 이 옵션의 설정을 체크하면 conflict 부분을 가시화 하는데 상당히 많은 메모리를 필요하게 되어 결과를 보는데 많이
시간이 필요하게 될 수 있습니다. 다음 옵션인 'Conflict resolution'은 conflict가 발생한 부분의 consensus 염기를 어떻게 결정할 것인지를 설정하는 옵션입니다. 그
리고 'Non-specific matches', 즉 reference 서열에 특정 부분에만 붙지 않고 다수의 부분에 붙는
read를 ‘Random'하게 붙일 것인지 ’Ignore'(무시) 할 것인지 설정 한 후 'Next'를 클릭합니다.
다음 단계에서 분석결과에 대한 report나 mapping 되지 않은 read들의 목록을 생성할 것인지, 결과를 저장할 것인지, 분석 로그를 생성시킬 것인지 등의 옵션을 설정합니다. 그 다음 단계에서 저장위치를 선택 후, 'Finish' 버튼을 클릭하면 reference assembly가 진행 됩니다.
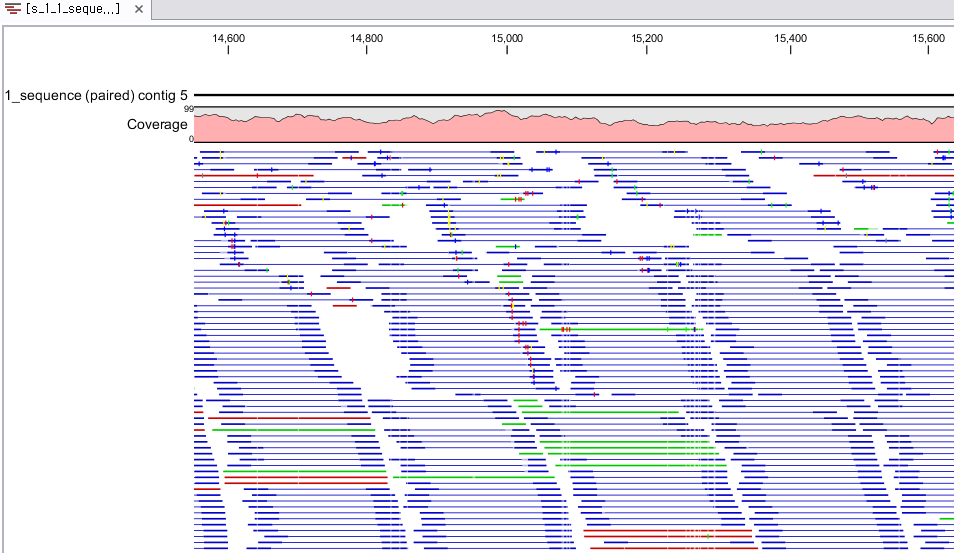
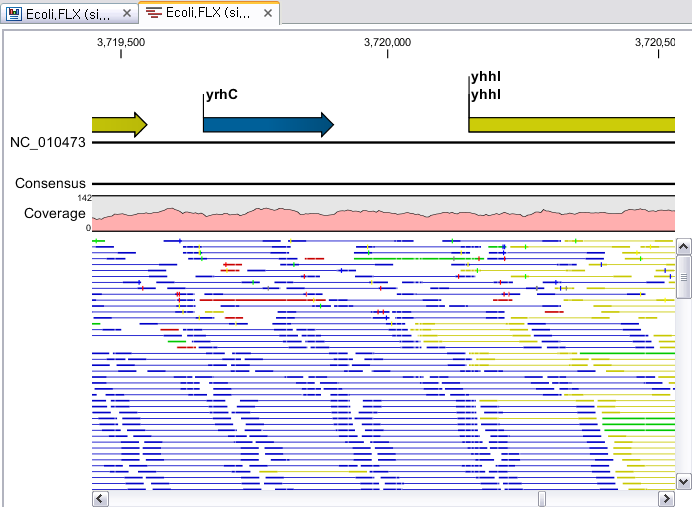
 Assembly가 완료되면 아래 그림처럼 mapping view가 나타나게 됩니다. 2개 이상의 reference 서열을 선택한
경우, 이전에 de novo assembly의 결과처럼 각 reference 서열 마다 간단한 정보와 함께 table 형태로
결과가 나타나게 되며, 각 항목을 더블클릭해 보면 mapping view를 보실 수 있습니다. de novo assembly
결과와는 다르게 consensus 서열 위에 reference 서열이 보이는 것을 보실 수 있습니다. 그리고 reference
서열에 있는 'Gene', 혹은 ‘CDS'와 같은 annotation들도 함께 보실 수 있습니다.
Assembly가 완료되면 아래 그림처럼 mapping view가 나타나게 됩니다. 2개 이상의 reference 서열을 선택한
경우, 이전에 de novo assembly의 결과처럼 각 reference 서열 마다 간단한 정보와 함께 table 형태로
결과가 나타나게 되며, 각 항목을 더블클릭해 보면 mapping view를 보실 수 있습니다. de novo assembly
결과와는 다르게 consensus 서열 위에 reference 서열이 보이는 것을 보실 수 있습니다. 그리고 reference
서열에 있는 'Gene', 혹은 ‘CDS'와 같은 annotation들도 함께 보실 수 있습니다.
* 아래 이메일 주소로 연락 주시면 CLC Genomics Workbench의 모든 기능을 사용할 수 있는 데모 라이센스를 제공해 드리오니 많은 이용 바랍니다.
- codes@insilicogen.com
어느 날 교수님께서 '너의 석사 주제를 가져왔노라'시며 의미심장한 미소를 띄우시고는 미팅룸으로 저를 끌고 가십니다.
요즘 NGS 라는게 뜨는데 네가 남자고 컴퓨터를 좀 더 잘 할 터이니 네가 이걸로 뭔가를 해보라고 하십니다.
그 '뭔가'가 대체 뭔지는 논문 찾아보면 다 나오니까 조사해오라고 하십니다.
나름 조사를 해보니 용어도 잘 모르겠고, 다 영어라 해석도 어렵고...
전 정말 미추어 버리겠지만, 어쩌겠습니까? 까라면 까야죠."
위의 이야기는 제가 아는 어떤 친구의 하소연입니다. 이런 상황은 이 친구 뿐만이 아니라 대한민국 실험실의 많은 학생들, 그리고 연구원들이 공감하는 이야기일 것 같습니다.
저도 그 답답함을 겪었던, 그리고 아직 겪고 있는 한 사람으로서 제가 아는 만큼 NGS에 대해서 쉽게 이해할 수 있는, 그리고 소통을 통해 그 답답함을 해결할 수 있게 하는 글을 남겨보고자 합니다. 자 그럼 NGS 정체에 대해서부터 이야기 해보겠습니다.
NGS가 뭡니까?
NGS는 Next Generation Sequencing의 약자로 차세대 염기서열 결정이라고 해석할 수 있겠습니다. 해석은 ‘염기서열 결정’이라고 했는데 ‘휴먼 지놈 시퀀싱’ 할 때의 그 시퀀싱을 쓰는게 더 어울릴 것 같네요. 아무튼 어떤 생명체의 염기 서열을 알아낼 때 쓰는 시퀀싱법의 최신 버젼을 말하는 것 같습니다. 차세대라고 하는걸 보면 이전 세대의 염기서열 결정법 보다 뭔가 더 좋은게 있는 것 같습니다. 얼마나 혁신적으로 좋아졌길래 ‘차세대’라는 말을 갖다 붙였을까요?
'이전 세대' 시퀀싱법 Sanger method
Sanger method는 학부 분자생물학 시간 때 언뜻 들은 기억이 납니다. 지놈을 무작위로 잘라 단편 조각으로 만들고, single strand로 만든 다음 PCR처럼 primer를 붙이고 중합효소로 상보적인 dNTP들을 붙여 나갈 때 형광다이가 붙어 있는 ddNTP가 붙으면 중합 반응이 끝나고, 그 조각들을 전기영동으로 분리하면 짧은 조각부터 1bp 간격으로 정렬이 되는데 이 순서대로 형광다이의 색으로 A, T, G, C 서열을 결정해 나가는 방법입니다. 비루한 저의 설명 보다는 없는 지식이 없는 위키피디아(http://en.wikipedia.org/wiki/DNA_sequencing)나 파워블로거 님들의 자료를 살펴 보심이 옳은 줄로 아뢰옵니다. Sanger method의 장점이라면 정확해서 믿을만하다고 할 수 있겠습니다. 단점은 비싸고 오래 걸린다는 점입니다. 그래서 연구용으로는 적합하지만 상업적 목적으로 이용하기에는 힘들다고 합니다.
그럼 NGS는 뭐가 좋은데?
싸고 빠릅니다. 어디까지나 Sanger method에 비해서라는 전제가 붙을 때 이야기입니다. 차세대 염기서열 결정법은 크게 4~5가지가 있습니다. 기존 Sanger method의 비싸고 오래 걸려서 상업적으로 이용하기 어렵다는 단점을 극복할 수 있는 시퀀싱법을 개발하기 위해 몇몇 회사가 뛰어들어 새로운 시퀀싱법을 개발해 냈는데 기본적으로 전체 genome을 잘라 단편으로 만든 후 각 단편의 서열을 시퀀싱하고 이 단편들을 assembly라는 단계에서 겹쳐지는 부분을 이어 원래 지놈 서열을 알아내는 식으로 genome project가 진행 됩니다. 각 회사마다 개발해낸 시퀀싱법이 적용된 장비(혹은 플랫폼이라고 부르기도 합니다.)를 개발해서 판매하고 있으며 대표적으로 Roche사의 GS-FLX, Illumina의 Genome Analyzer, Applied Biosystem사의 SOLiD, Life Technologies사의 Ion Torrent 등이 있습니다. 여기서부터 슬슬 복잡해지기 시작합니다. 각 장비들마다 다른 시퀀싱법을 사용하다 보니 저마다 각자의 강점과 약점이 있습니다.
NGS로 무엇을 할 수 있을까?
서열을 시퀀싱해서 할 수 있는 분석들 대부분을 할 수 있습니다. 크게 나누자면 genomics, transcriptomics, epigenomics 분야로 나눌 수 있지만 raw data를 어떻게 응용하느냐에 따라 더 다양한 분야의 분석 방법들이 만들어 질 수 있을 것 같습니다.
Genomics | Transcriptomics | Epigenomics |
de novo assembly Reference assembly SNP detection DIP detection | RNA-Seq Small-RNA analysis | ChIP-Seq |
NGS 분석을 위해 준비해야 할 것들
"자 그럼 NGS 분석을 해보자! 뭐부터? 설계 먼저!
헐~ 데이터 파일이 너무 큰데... 내 PC에서 하면 PC 폭발하는거 아녀?
받긴 받았는데.. 이게 다 뭐다냐?"
어느 실험이 다 그렇듯이 NGS 데이터를 분석하는 것도 정확한 계획을 세우는 것이 중요합니다. 그 계획을 세우기 위해서는 시퀀싱 대상이 원핵 생물인지 진핵 생물인지? 지놈의 크기는 어느 정도인지? 그리고 얼마나 많이 시퀀싱 할 것인지(depth)? 그리고 주로 분석 할 대상이 RNA서열인지? exom 서열인지? whole 지놈 서열인지? 와 같이 시퀀싱 대상에 대한 정보를 파악하고 분석 목적에 맞는 시퀀싱 데이터(single short, long read, paired-end read, mate paired-end read)를 만드는 것이 중요합니다. 그리고 이 데이터들을 분석하기 위한 고사양의 컴퓨터, 분석 소프트웨어도 준비해야 합니다. 이 부분에 대한 설명은 다음으로 넘기도록 하겠습니다.
"de novo" assembly를 해보자!
'de novo' 라는 말이 낯섭니다. 우리 친구 네이놈 사전을 찾아보니 부사로써 ’처음부터‘, ’새로(이)(anew)‘, ’다시(again)‘, 영영사전에는 'from the beginning' 이라는 뜻이랍니다. 그러니까 de novo assembly는 새롭게 assembly를 한다는 말이 되겠네요. de novo assembly는 주로 서열이 밝혀지지 않은 종의 지놈 서열을 알아내고자 할 때 수행하는 분석입니다. Assembly는 일종의 퍼즐 맞추기랑 비슷합니다. Reference assembly는 원본 사진을 보면서 맞추는 퍼즐이고 de novo assembly는 원본 사진 없이 맞추는 거라고 보시면 됩니다.
원본 없이 하나하나 맞춰보면서 연결되는 것들 끼리 쭉~ 이어 원래의 genome 서열을 알아내야 하는 작업인지라 한 종류의 read들만 가지고 assemble을 하기란 쉽지 않습니다. 그래서 short, long, paired-end, mate-paired-end 등 여러 종류의 read들을 가지고 assembly를 해야 결과가 좋아집니다.
NGS read 데이터를 다운받자!
Assembly를 하려면 먼저 NGS read가 필요합니다. 현재 NCBI, EBI, DDBJ의 sequence 데이터 저장 서비스인 SRA(Sequence Read Archive)에서 공개된 NGS 데이터를 받을 수 있습니다. 일단 이 블로그에서는 어떻게 de novo assembly를 돌릴 수 있는지 알아보는 것이 목적이므로 지놈의 크기가 비교적 작은 E. coli의 NGS 데이터를 사용하고자 합니다. 웹 브라우저에서 아래 url로 이동해 보시면 CLC bio에서 제공하는 예제 데이터로 E.coli의 genome을 sequencing한, Roche의 454 장비에서 만들어진 long read 데이터와, Illumina 장비에서 만들어진 paired-end read 데이터를 다운받을 수 있습니다.
http://www.clcbio.com/index.php?id=1290
Roche 454 데이터 불러오기
Roche의 데이터는 크게 두 가지 형태가 있습니다. 하나는 454 Flowgram 이라고 불리는 .sff 파일이고 다른 하나는 FASTA 포맷으로 서열과 그에 대응하는 quality 값이 각각 두 개의 파일에 분리된 형태가 있습니다. 여기서 사용할 데이터는 후자에 해당합니다. 먼저 다운로드 받은 Roche 데이터의 압축 파일을 풀면 3개의 파일이 생성됩니다. 하나는 이미 알려진 E.coli의 전체 genome 서열 파일(.gbk)이고 다른 두 파일에 NGS read 파일입니다. 이중에서 .fna 파일이 각 read의 서열이 있는 파일이고 .qual 파일은 각 서열의 quality 값이 순서대로 기록된 파일입니다. 그 리고 CLC Genomics Workbench 화면 위쪽의 Tool bar에서 'NGS import'라는 버튼을 클릭하면 다음과 같이 import 할 수 있는 NGS data의 종류가 나타납니다. 여기서 첫 번째에 있는 'Roche 454...‘를 클릭합니다.
Illumina 데이터 불러오기
이번에 불러올 Illumina의 데이터는 paired-end read입니다. 검색위치에서 Illumina 데이터의 압축을 풀어놓은 폴더를 찾아간 후 forward 방향의 read 파일과 reverse 방향의 read파일을 둘다 선택 하시고, ‘General options'에 있는 ’Paired reads'라는 옵션을 체크해 줘서 이 read들이 paired-end read임을 인식하도록 해줍니다. 'Paired read' 옵션이 선택되면 ‘Paired read orientation'이라는 항목에 대한 옵션을 조정 할 수 있도록 활성화가 되는데 여기서 insertion size를 조정해 줍니다. 그리고 ’Next' 버튼을 클릭하고 저장할 위치를 설정한 후 ‘Finish' 버튼을 클릭하면 import가 완료됩니다.
de novo assembly 돌리기
Import가 완료되면 다음 그림과 같이 Roche와 Illumina 데이터가 각각 하나씩 나타나게 됩니다. Illumina 데이터의 파일은 두 개였지만 import 가 되면서 하나의 데이터로 합쳐진 것을 볼 수 있습니다.
Reference assembly를 해보자!
앞에서 reference assembly가 원본 사진을 보고 퍼즐을 맞추는 것과 비슷하다고 말했습니다. Reference assembly는 말 그대로 reference가 되는 원본 서열에 read들을 mapping시켜 만들어지는 consensus 서열을 얻는 것을 말합니다. 예를 들어 한우의 유전체를 시퀀싱 하여 얻어낸 read 데이터를 NCBI에 공개된 소의 reference 서열에 mapping 시켜서 한우의 유전체와 어떤 차이가 있는지 비교하는 분석을 수행할 수 있을 것입니다.
이번에도 Roche 454 데이터와 Illumina의 데이터를 이용하되, 함께 압축되어 있던 E.coli 지놈 서열(NC_010473)을 reference로 두고 mapping 시켜 보도록 하겠습니다. Reference 서열 파일, NC_010473.gbk는 마우스로 클릭앤드래그하여 ‘Navigation Area’에 옮기면 자동으로 import가 완료 됩니다.
다음 단계에서 분석결과에 대한 report나 mapping 되지 않은 read들의 목록을 생성할 것인지, 결과를 저장할 것인지, 분석 로그를 생성시킬 것인지 등의 옵션을 설정합니다. 그 다음 단계에서 저장위치를 선택 후, 'Finish' 버튼을 클릭하면 reference assembly가 진행 됩니다.
* 아래 이메일 주소로 연락 주시면 CLC Genomics Workbench의 모든 기능을 사용할 수 있는 데모 라이센스를 제공해 드리오니 많은 이용 바랍니다.
- codes@insilicogen.com
Posted by 人Co
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/106
Trackback URL : 이 글에는 트랙백을 보낼 수 없습니다

